The Site-Specific Glycosylation Challenge
A single monoclonal antibody carries one N-glycosylation site on each heavy chain — two per intact IgG. That seems manageable. But human plasma contains over 500 confirmed N-glycosylation sites distributed across more than 150 glycoproteins, and each site can be occupied by any of several hundred distinct glycan structures. The same protein can carry different glycans at the same site in different tissues, different disease states, and different individuals. Multiply those variables and you arrive at the central problem of glycoproteomics: glycosylation is combinatorial, and site specificity is the only biologically meaningful unit of measurement. For additional insights into glycoproteomics shipping checklist, explore our in-depth resource.
Knowing that a glycoprotein is "fucosylated" tells you almost nothing. Fucosylation of the core N-acetylglucosamine on IgG1 at Asn297 blocks antibody-dependent cellular cytotoxicity. Fucosylation of an antennary GlcNAc on the same glycan has no such effect. Fucosylation of an O-glycan on a mucin domain in MUC16 modulates cancer cell adhesion through an entirely different mechanism. The same monosaccharide, the same modification, three different biological outcomes — because the site and structural context differ. Glycoproteomics, as distinct from glycomics (which analyzes released glycans) and glycoprotein analysis (which characterizes glycosylation at the whole-protein level), is the discipline that maps exactly which glycan occupies which amino acid on which protein in a given sample.
This article provides a practical, enrichment-to-software guide for glycoproteomics by LC-MS/MS. For researchers evaluating glycoproteomics capabilities, Creative Proteomics provides protein glycosylation analysis services spanning both glycomics and glycoproteomics workflows.
Sample Preparation for Complex Biospecimens
The site-specific glycosylation profile of a protein is exquisitely sensitive to sample handling. Glycosidases released during tissue homogenization can trim terminal sialic acids and fucose residues within minutes post-lysis. Endogenous PNGase F-like activities in some tissues can release entire N-glycan chains. The sample preparation protocol must achieve three things simultaneously: protein denaturation to inactivate glycosidases, preservation of labile glycan modifications (particularly O-acetylation and sulfation), and compatibility with downstream enrichment chemistry.
For plasma and serum, the primary challenge is dynamic range. Albumin and IgG together account for over 65% of plasma protein mass, obscuring low-abundance glycoproteins that may carry the most informative glycosylation changes. Immunodepletion of the top 7 or top 14 abundant proteins is standard practice before glycoproteomic analysis, though depletion columns introduce their own biases — some low-abundance glycoproteins co-deplete through protein-protein interactions. An alternative approach uses glycopeptide-level enrichment directly from undepleted plasma digests, relying on the enrichment chemistry to separate glycopeptides from non-glycosylated peptides rather than depleting proteins upfront. This glycopeptide-first strategy avoids immunodepletion bias but requires higher enrichment specificity.
For tissue samples, snap-freezing in liquid nitrogen is the minimum standard. Formalin-fixed paraffin-embedded (FFPE) tissue — the dominant archival format in clinical research — presents a particular challenge: formalin crosslinking masks glycosylation sites, and heat-induced antigen retrieval can partially deglycosylate proteins. PNGase F digestion from FFPE sections enables N-glycan analysis with acceptable reproducibility, though intact glycopeptide analysis from FFPE yields substantially lower coverage than from frozen tissue, and site occupancy information is lost when glycans are released rather than analyzed as intact glycopeptides.
 Figure 1: Complete glycoproteomics workflow flowchart — from sample types (plasma, tissue biopsy, cell pellet, FFPE section) through preparation (immunodepletion vs. direct digestion), trypsin digestion, enrichment strategy selection, LC-MS/MS acquisition mode selection, and software analysis pipelines, to biological interpretation (site occupancy, glycan structure, differential abundance).
Figure 1: Complete glycoproteomics workflow flowchart — from sample types (plasma, tissue biopsy, cell pellet, FFPE section) through preparation (immunodepletion vs. direct digestion), trypsin digestion, enrichment strategy selection, LC-MS/MS acquisition mode selection, and software analysis pipelines, to biological interpretation (site occupancy, glycan structure, differential abundance).
For clinical cohort studies where samples have been collected over months or years with varying pre-analytical conditions, standardized sample preparation protocols are essential to minimize pre-analytical variability in glycoproteomic measurements.
Glycopeptide Enrichment: Choosing the Right Chemistry
Enrichment is the step that determines which subpopulation of glycopeptides enters the mass spectrometer. No single enrichment method captures all glycopeptide classes with equal efficiency, and the choice of method — or combination of methods — must align with the biological question.
Hydrophilic Interaction Liquid Chromatography (HILIC) is the most widely used enrichment mode for intact glycopeptide analysis and the closest thing to an "unbiased" method. The principle is straightforward: the glycan moiety is markedly more hydrophilic than the peptide backbone, so glycopeptides are retained on a polar stationary phase (amide, zwitterionic, or maltose-functionalized silica) under high organic conditions (70-80% acetonitrile) while non-glycosylated peptides are eluted.
ZIC-HILIC (zwitterionic HILIC) using sulfobetaine-functionalized silica provides consistently high specificity in comparative studies. A 2026 study in Talanta demonstrated that reducing the loading ACN from 80% to 70% on HILIC-Amide columns captured an additional 20% of highly hydrophilic glycopeptides missed by conventional protocols, primarily those carrying large, multi-antennary sialylated glycans. The complementary 70% and 80% ACN fractions together provide broader coverage than either condition alone.
Lectin affinity chromatography exploits the natural specificity of plant and fungal lectins for particular glycan structural features. Concanavalin A (ConA) binds high-mannose and hybrid N-glycans; wheat germ agglutinin (WGA) binds sialic acid and GlcNAc; Aleuria aurantia lectin (AAL) binds fucose; Sambucus nigra agglutinin (SNA) preferentially binds α2,6-linked sialic acid. Multi-lectin columns combining ConA, WGA, and Jacalin (for O-glycans) provide broader specificity than single-lectin columns. Lectin enrichment excels when the biological question is structure-specific — for instance, enriching fucosylated glycopeptides from hepatocellular carcinoma serum where increased core fucosylation is a known biomarker. The limitation is inherent bias: you only find what the lectin recognizes.
Boronic acid affinity uses the reversible covalent interaction between boronate and cis-diol groups present on most monosaccharides (mannose, galactose, fucose, GlcNAc in its open-ring form). Phenylboronic acid-functionalized magnetic beads or monolithic columns capture glycopeptides at alkaline pH (8-9) and release them at acidic pH. Boronic acid enriches all classes of glycosylation and is considerably cheaper than lectin columns. The limitation is lower specificity than HILIC — non-glycosylated peptides containing serine and threonine-rich sequences show nonspecific binding — and sialic acids, which lack a free cis-diol, are captured less efficiently.
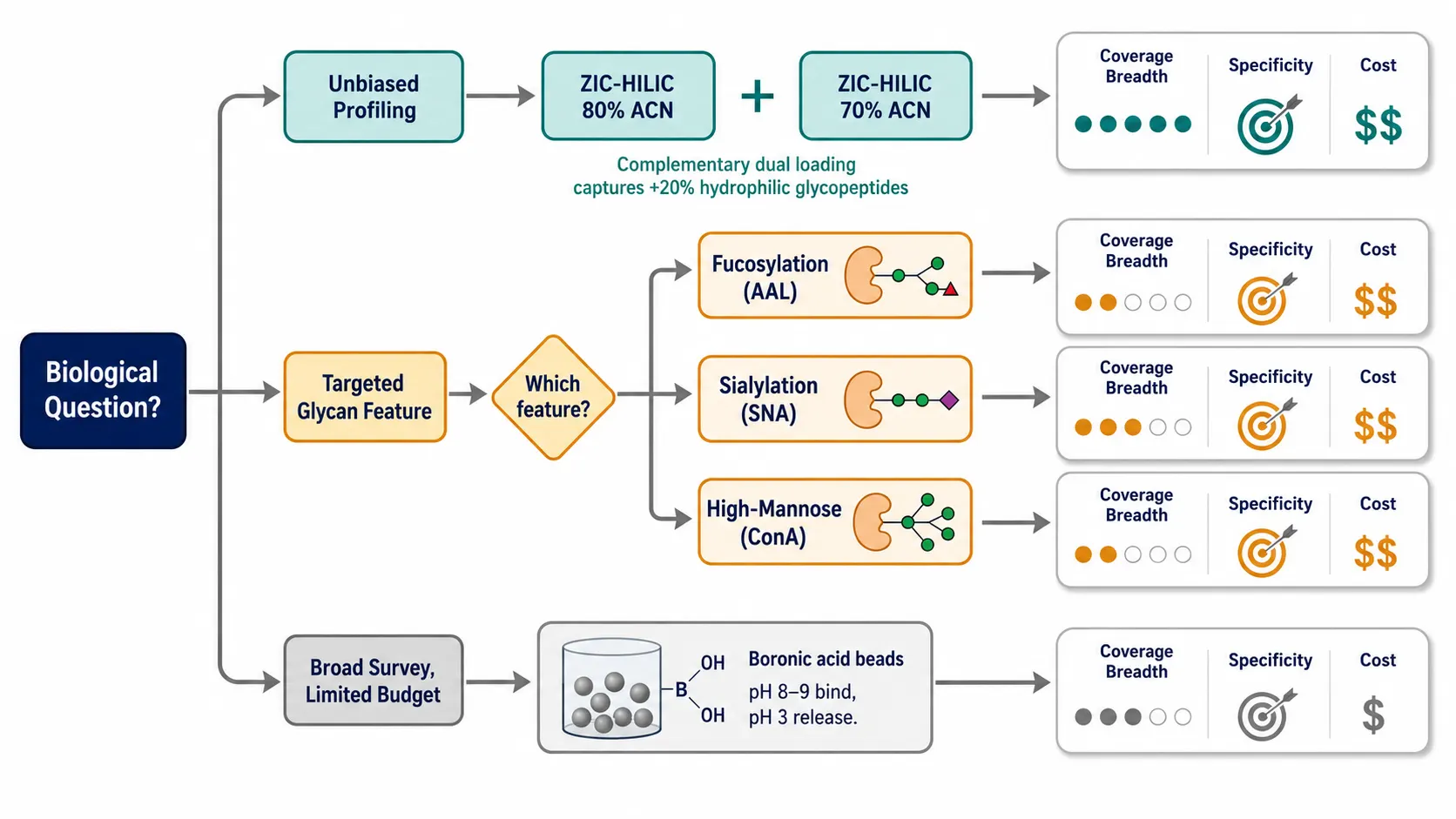 Figure 2: Decision tree for glycopeptide enrichment method selection — starting question: "What is your biological question?" branching into three tracks: (A) "Unbiased profiling" → ZIC-HILIC with complementary 70%/80% ACN dual loading; (B) "Targeted glycan feature (e.g., fucosylation, sialylation)" → specific lectin or multi-lectin column matching the structural feature; (C) "Broad glycopeptide survey on a budget" → boronic acid affinity. Each terminal node annotated with coverage breadth, specificity, equipment requirements, and recommended protocol reference.
Figure 2: Decision tree for glycopeptide enrichment method selection — starting question: "What is your biological question?" branching into three tracks: (A) "Unbiased profiling" → ZIC-HILIC with complementary 70%/80% ACN dual loading; (B) "Targeted glycan feature (e.g., fucosylation, sialylation)" → specific lectin or multi-lectin column matching the structural feature; (C) "Broad glycopeptide survey on a budget" → boronic acid affinity. Each terminal node annotated with coverage breadth, specificity, equipment requirements, and recommended protocol reference.
Mixed-mode approaches are increasingly adopted in large-scale studies. A ZIC-HILIC enrichment followed by a second-dimension separation using high-pH reversed-phase fractionation can increase glycopeptide identifications by 30-40% over ZIC-HILIC alone. For studies prioritizing depth over throughput, two-dimensional enrichment — HILIC followed by lectin affinity on the HILIC eluate — provides both breadth and structural selectivity.
For researchers designing glycoproteomics experiments, Creative Proteomics provides glycopeptide analysis services with method development across HILIC, lectin, and mixed-mode enrichment strategies, and comprehensive glycomics services covering both released glycan analysis and intact glycopeptide workflows.
LC-MS/MS Acquisition Strategies
Enrichment delivers glycopeptides to the LC-MS. The acquisition method determines whether you can assign both the peptide sequence and the glycan composition to the same precursor ion.
Data-dependent acquisition (DDA) remains the workhorse for discovery glycoproteomics. The mass spectrometer selects the most abundant precursor ions from each MS1 scan for fragmentation. For glycopeptides, stepped HCD — applying three collision energies (typically 25, 35, and 45 normalized collision energy) to the same precursor — has become the standard approach. The lower energy fragments the glycan (producing diagnostic oxonium ions at m/z 204.0872 for HexNAc, m/z 366.140 for HexNAc-Hex, and m/z 292.103 for sialic acid), while the higher energy fragments the peptide backbone for sequence identification. A 2024 systematic evaluation in Analytical Chemistry demonstrated that ZIC-HILIC enrichment combined with stepped-HCD DDA and TMT quantification identified over 5,500 unique intact glycopeptides from cell lysate, with an average CV of 8% for TMT-based quantification.
EThcD (electron transfer with supplemental HCD) is the preferred fragmentation mode for O-glycopeptides. O-linked glycans are more labile than N-linked glycans under collisional activation, and ETD preserves the glycan while fragmenting the peptide backbone. The supplemental HCD generates glycan oxonium ions and peptide b/y-ions, providing complementary information to the ETD c/z-ion series. On Orbitrap Tribrid instruments, EThcD is recommended whenever O-glycopeptide identification is a study objective.
Data-independent acquisition (DIA) is gaining traction in glycoproteomics. Rather than selecting individual precursors, DIA fragments all ions within sequential isolation windows, generating a complete fragment ion map of the sample. The advantage is reproducibility — every precursor is fragmented in every run, eliminating the stochastic precursor selection that makes DDA irreproducible across replicates. The challenge is computational: deconvoluting glycopeptide spectra from DIA data requires software that can simultaneously assign peptide sequence, glycan composition, and site localization from multiplexed spectra. DIA-based glycoproteomics using timsTOF instruments with parallel accumulation-serial fragmentation (PASEF) can identify 3,000-4,000 glycopeptides from a single 90-minute gradient, competitive with optimized DDA.
PRM (parallel reaction monitoring) and targeted approaches are used for validation. Once candidate glycopeptide biomarkers are identified by DDA or DIA, PRM assays targeting the specific precursor m/z and monitoring a panel of diagnostic fragment ions (peptide backbone fragments + glycan oxonium ions) provide quantitative confirmation with higher sensitivity and specificity than discovery-mode acquisition.
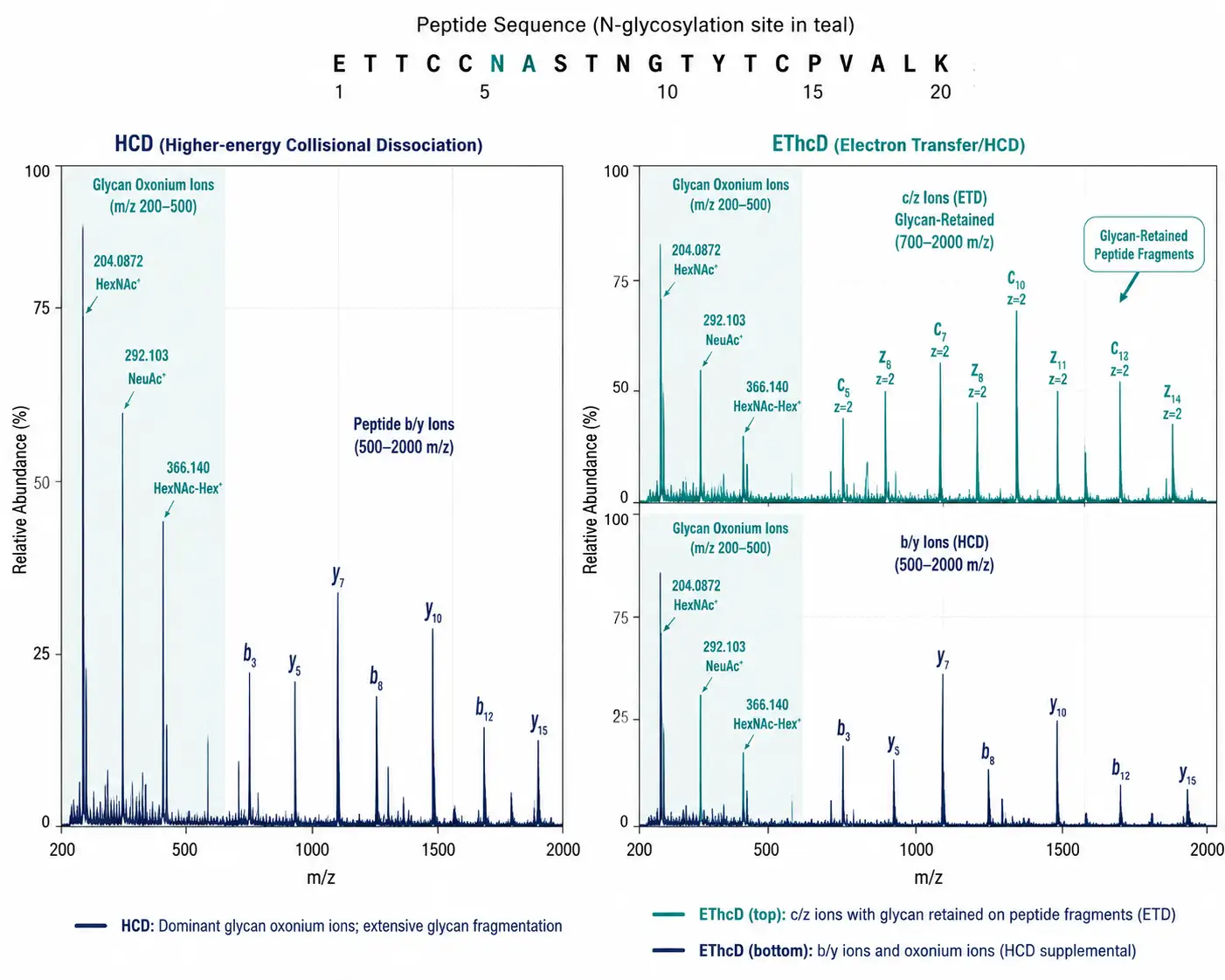 Figure 3: Comparison spectra layout — left panel showing an HCD spectrum of an intact N-glycopeptide with dominant oxonium ions (m/z 204, 366) and peptide b/y fragments; right panel showing an EThcD spectrum of the same glycopeptide with c/z ions from ETD (glycan retained on fragments) supplemented by b/y ions and oxonium ions from HCD. Annotations highlight the diagnostic value of each fragmentation mode.
Figure 3: Comparison spectra layout — left panel showing an HCD spectrum of an intact N-glycopeptide with dominant oxonium ions (m/z 204, 366) and peptide b/y fragments; right panel showing an EThcD spectrum of the same glycopeptide with c/z ions from ETD (glycan retained on fragments) supplemented by b/y ions and oxonium ions from HCD. Annotations highlight the diagnostic value of each fragmentation mode.
For instrument selection, the key trade-off is between resolution, speed, and ETD capability. Orbitrap Tribrid instruments (Fusion Lumos, Eclipse) offer EThcD for O-glycoproteomics and resolution up to 500,000 for accurate mass measurement. The timsTOF platform provides ion mobility separation (CCS values as an additional dimension for glycopeptide identification) and high acquisition speed for DIA-PASEF workflows. Q-TOF instruments offer robust performance at lower capital cost but lack ETD capability for O-glycopeptide characterization. For deep quantitative glycoproteomics, high-resolution MS/MS with stepped-HCD and TMT-based quantification provides reproducible site-specific glycan occupancy data across large sample cohorts.
Data Analysis: Software Tools and Validation
Glycoproteomics data analysis is computationally distinct from standard proteomics. A proteomics database search tests the hypothesis "this MS/MS spectrum comes from peptide X with modification Y." A glycoproteomics search tests "this MS/MS spectrum comes from peptide X with glycan Z," where Z is drawn from a glycan database containing hundreds to thousands of possible compositions. The search space explosion requires software purpose-built for glycopeptide assignment.
A 2025 comparative evaluation published in Analytical and Bioanalytical Chemistry benchmarked five glycoproteomics search engines — Byonic, Protein Prospector, MSFragger-Glyco, pGlyco3, and GlycoDecipher — against a tailored glycan database. The key finding was sobering: no single tool was a clear winner across all criteria, and Byonic — the most widely used commercial glycoproteomics software — produced an elevated rate of spurious glycoprotein and glycosite assignments compared to the other four tools. A separate 2025 bioRxiv study comparing the same tools found that approximately 50% of identified glycopeptides were unique to individual software tools, with Byonic identifying the most total glycopeptides but MSFragger-Glyco showing the highest rate (16.6%) of identifications lacking the diagnostic Y0 or Y1 ions that confirm the glycan assignment is based on glycan-specific fragments rather than noise.
The practical implication is that glycoproteomics data should be processed with at least two complementary search engines — one peptide-first (MSFragger-Glyco or Protein Prospector) and one glycan-first (pGlyco3 or GlycoDecipher) — and only glycopeptides identified by both should be treated as high-confidence. Additional filtering on diagnostic ion presence (Y0 or Y1 ion with intensity >1% of base peak) and glycan composition plausibility further reduces false assignments.
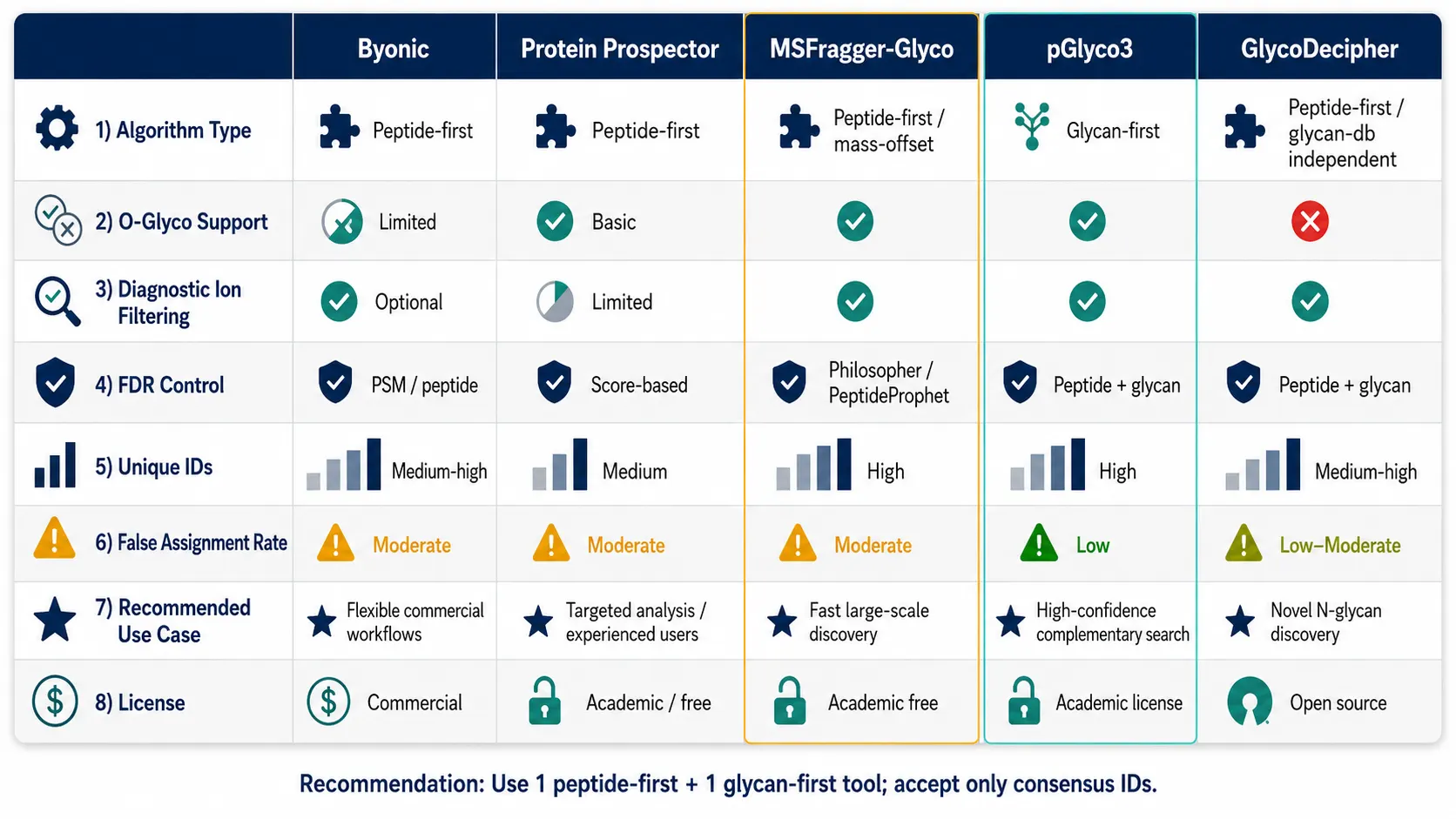 Figure 4: Software comparison matrix — 5 columns (Byonic, Protein Prospector, MSFragger-Glyco, pGlyco3, GlycoDecipher) × rows covering: search algorithm type (peptide-first vs glycan-first), O-glyco support, diagnostic ion filtering, FDR control method, unique glycopeptide IDs in benchmark dataset, false positive rate estimate, recommended use case, and license type (commercial vs. open-source).
Figure 4: Software comparison matrix — 5 columns (Byonic, Protein Prospector, MSFragger-Glyco, pGlyco3, GlycoDecipher) × rows covering: search algorithm type (peptide-first vs glycan-first), O-glyco support, diagnostic ion filtering, FDR control method, unique glycopeptide IDs in benchmark dataset, false positive rate estimate, recommended use case, and license type (commercial vs. open-source).
For quantitative glycoproteomics data, Creative Proteomics provides bioinformatics support for proteomics including glycopeptide-specific database search configuration and multi-tool consensus analysis pipelines.
N-Glycoproteomics vs. O-Glycoproteomics: Distinct Workflows
N-linked and O-linked glycosylation present fundamentally different analytical challenges, and a workflow optimized for one will perform poorly on the other.
N-glycoproteomics benefits from a conserved sequon (Asn-X-Ser/Thr, X ≠ Pro) that allows in silico prediction of glycosylation sites from the protein sequence. The N-glycan core structure (GlcNAc₂Man₃) is conserved across all eukaryotes, and PNGase F — a bacterial enzyme that cleaves the bond between the innermost GlcNAc and the asparagine side chain — specifically and quantitatively releases N-glycans. PNGase F digestion in H₂¹⁸O labels the deamidated asparagine (Asn→Asp + 3 Da mass shift), providing a chemical marker for former N-glycosylation sites. This deglycosylation-based workflow — release glycans with PNGase F, analyze released glycans separately, identify deamidated sites by proteomics — is technically simpler than intact glycopeptide analysis and remains common for N-glycosylation site mapping. The limitation is that site-specific glycan occupancy information is lost: you know which sites are glycosylated but not which glycan occupied which site.
O-glycoproteomics has no universal enzyme equivalent to PNGase F. O-glycans lack a conserved core structure (though mucin-type O-glycans share a GalNAc-α-Ser/Thr core), there is no consensus sequon for O-glycosylation, and no enzyme cleaves all O-glycan types quantitatively. O-glycoproteomics must therefore be performed on intact O-glycopeptides, which requires EThcD fragmentation to preserve the labile O-glycan during peptide backbone fragmentation, specialized enrichment (Jacalin lectin for mucin-type O-glycans, or HILIC with optimized conditions), and software capable of searching for O-glycan modifications without a sequon constraint.
For studies requiring site-specific O-glycosylation analysis, Creative Proteomics provides O-glycosylation site occupation analysis and O-glycan profiling services with EThcD-enabled workflows. For N-glycosylation characterization, N-glycosylation site occupation analysis and N-glycan profiling cover both released glycan and intact glycopeptide approaches.
Case Studies in Site-Specific Glycosylation
The SugarQuant approach, published by the Heck laboratory, exemplifies a state-of-the-art quantitative N-glycoproteomics workflow. Peptides are labeled with TMTpro 16-plex, enriched by ZIC-HILIC, and analyzed by stepped-HCD on an Orbitrap Eclipse. The TMT reporter ions in the low-mass region provide relative quantification across 16 samples, while the stepped-HCD fragmentation provides glycan composition from oxonium ions and peptide sequence from b/y-ions. Applied to a panel of breast cancer cell lines, SugarQuant quantified over 3,000 unique intact glycopeptides and identified site-specific fucosylation changes on integrins and growth factor receptors associated with the mesenchymal phenotype. The TMT multiplexing is critical: it allows 16 samples to be compared in a single experiment, eliminating run-to-run variation in glycopeptide enrichment and LC separation.
Mucin-domain glycoproteomics represents the frontier of O-glycoproteomics. Mucins such as MUC1, MUC4, and MUC16 contain tandem repeat domains densely decorated with O-GalNAc glycans — up to 80% of the molecular weight of MUC16 is carbohydrate. Mapping site-specific O-glycosylation on mucins requires a specialized workflow: the mucin domain is enriched by Jacalin or Vicia villosa agglutinin, digested with a panel of proteases (trypsin alone cannot cleave within the densely glycosylated tandem repeats), and analyzed by EThcD to preserve the O-glycan during peptide fragmentation. The emerging picture from mucin glycoproteomics is that glycosylation patterns are not random: specific Ser/Thr residues within the tandem repeat are preferentially glycosylated, and the pattern of occupancy — which residues are glycosylated, not just which glycans are attached — determines mucin interactions with galectins and siglecs.
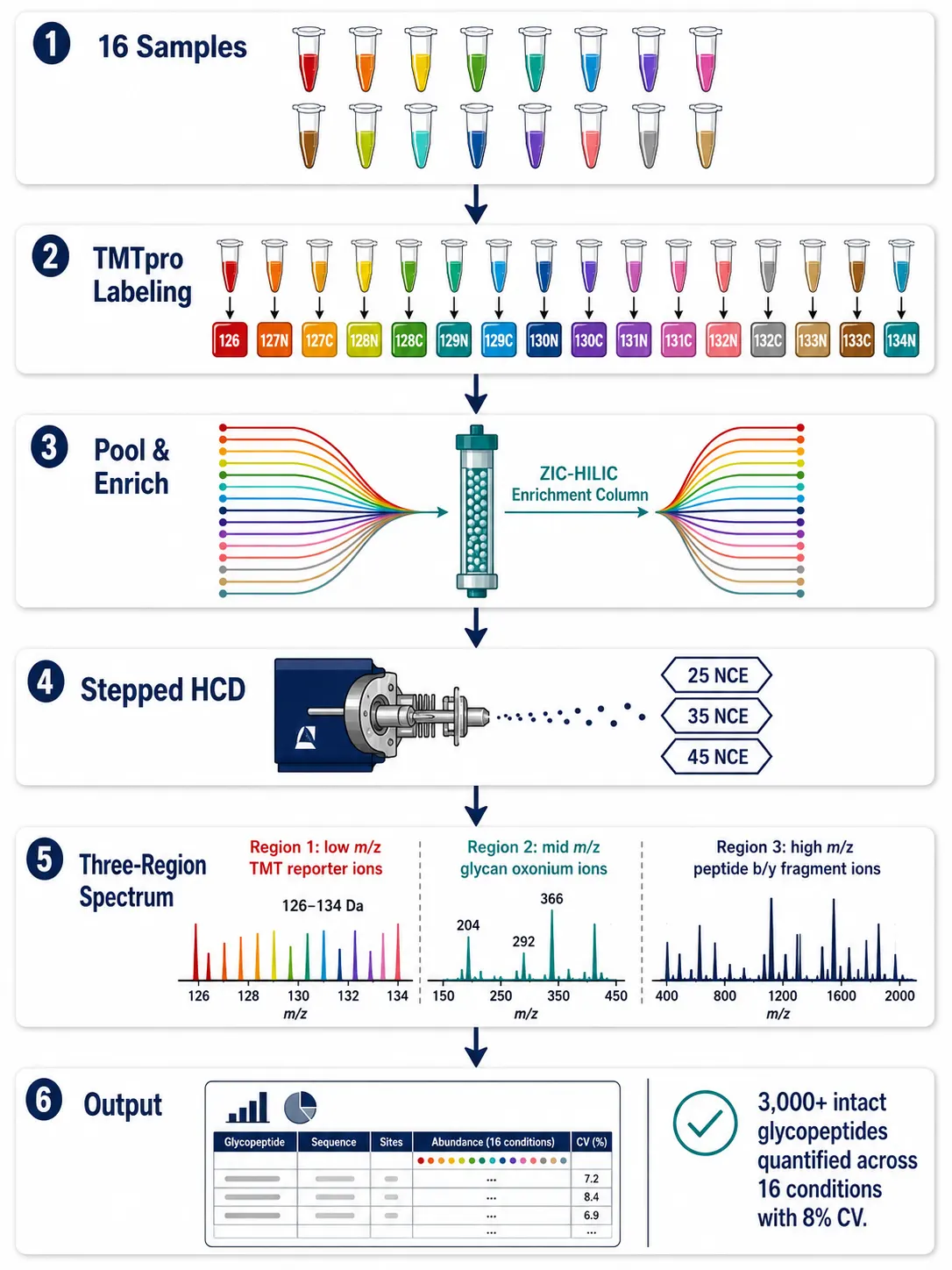 Figure 5: SugarQuant TMT-based quantitative glycoproteomics workflow diagram — 16-plex TMT labeling → ZIC-HILIC enrichment → stepped-HCD on Orbitrap Eclipse → TMT reporter ion quantification (low m/z) + glycan oxonium ion profiling (mid m/z) + peptide sequencing (high m/z) → site-specific glycan occupancy quantification across conditions.
Figure 5: SugarQuant TMT-based quantitative glycoproteomics workflow diagram — 16-plex TMT labeling → ZIC-HILIC enrichment → stepped-HCD on Orbitrap Eclipse → TMT reporter ion quantification (low m/z) + glycan oxonium ion profiling (mid m/z) + peptide sequencing (high m/z) → site-specific glycan occupancy quantification across conditions.
For researchers investigating specific glycan structural features in biological contexts, complementary techniques such as glycan microarray assays enable high-throughput screening of glycan-protein interactions — see our guide on glycan microarray technology for host-pathogen interaction studies. For structural characterization of complex polysaccharides, see our article on polysaccharide structural analysis from monosaccharide composition to linkage characterization.
Emerging Technologies
Trapped ion mobility spectrometry (timsTOF) provides collision cross section (CCS) values as a fourth dimension of separation (retention time, m/z, ion intensity, CCS). For glycopeptides, CCS values correlate with glycan structural features — a bisecting GlcNAc or a core fucose shifts the CCS measurably, even when the glycan composition (and thus mass) is identical. This means that isomeric glycopeptides, which are indistinguishable by mass alone, can potentially be resolved by ion mobility. The timsTOF platform combined with DIA-PASEF acquisition is currently generating the largest published glycoproteomic datasets, and glycan-specific CCS libraries are being built to support isomer-resolved glycoproteomics.
Artificial intelligence and deep learning are beginning to address three persistent bottlenecks in glycoproteomics. First, multi-task deep learning models can now predict fucosylation status, glycan class, and branching type directly from MS/MS spectra with over 96% accuracy, as demonstrated at HUPO 2025. Second, transformer-based models trained on glycoproteomics data can predict glycosylation site occupancy from primary sequence alone — not just the presence of a sequon, but whether a specific Asn or Ser/Thr in a specific protein context is likely to be occupied. Third, AI-driven spectral prediction for glycopeptides (analogous to Prosit for unmodified peptides) is in active development, which would allow spectral library searching for glycoproteomics and dramatically improve DIA data analysis.
Single-cell glycoproteomics remains a frontier goal: current workflows require microgram quantities of protein (approximately 100,000-500,000 cells), orders of magnitude above single-cell inputs. Nanodroplet sample processing and ultra-low-flow LC-MS are pushing toward single-cell proteomics; glycoproteomics will follow as enrichment sensitivity improves.
Figure 6: Emerging technologies landscape — three-panel horizontal layout showing (left) timsTOF CCS-based isomer separation with a schematic of isomeric glycopeptides resolved by ion mobility, (center) AI/deep learning pipeline from MS/MS spectrum input to glycan class/fucosylation/branching prediction output, (right) single-cell glycoproteomics concept showing nanodroplet processing and ultra-low-flow LC-MS.
For researchers investigating mucin-type O-glycosylation in cancer or other disease contexts, see our guide on O-glycan and N-glycan profiling workflows for mucin and glycoprotein research.
FAQ
What is the difference between glycomics and glycoproteomics?
Glycomics analyzes glycans after releasing them from proteins (typically with PNGase F for N-glycans or β-elimination for O-glycans), providing glycan composition and structure but losing the information about which glycan was attached to which protein at which site. Glycoproteomics analyzes intact glycopeptides — the glycan still attached to its peptide — preserving site-specific occupancy information.
Which enrichment method is best for glycoproteomics?
ZIC-HILIC is the closest to an unbiased method and the recommended starting point for discovery glycoproteomics. Lectin affinity is preferred for targeted enrichment of specific glycan structural features (fucosylation, sialylation). Boronic acid affinity offers broad coverage at lower cost but with reduced specificity. Using complementary 70% and 80% ACN HILIC loading conditions captures a broader range of glycopeptide hydrophilicity.
Can I analyze N-glycoproteomics and O-glycoproteomics in the same experiment?
Yes, but with compromises. Click-iG and similar methods use metabolic labeling to enrich N- and O-glycopeptides simultaneously. However, O-glycopeptides require EThcD for reliable identification, and EThcD is slower than HCD. A combined experiment on an Orbitrap Tribrid using EThcD for all precursors will identify both N- and O-glycopeptides but at lower throughput than dedicated HCD-only N-glycoproteomics. For studies requiring deep coverage of both, separate N-glyco and O-glyco experiments with optimized methods for each are recommended.
What is the minimum sample amount for glycoproteomics?
For cell lysate, approximately 100-500 μg of protein (roughly 1-5 million cells) is sufficient for a standard ZIC-HILIC enrichment and single-shot LC-MS/MS analysis identifying 2,000-4,000 intact glycopeptides. For plasma, 10-50 μL after depletion of abundant proteins is typical. Tissue samples require 5-20 mg wet weight depending on tissue type. These amounts assume optimized sample preparation; suboptimal handling increases the required input.
How reliable are glycoproteomics software tools?
A 2025 multi-tool benchmark found that no single software tool is sufficient — approximately 50% of glycopeptide identifications are unique to individual tools. The recommended approach is to process data with at least two complementary search engines (one peptide-first, one glycan-first) and accept only glycopeptides identified by both. Byonic, despite being the most widely used, showed elevated false discovery rates in multiple 2025 comparisons.
Why is EThcD preferred for O-glycoproteomics?
O-linked glycans are more labile than N-linked glycans under collisional activation and are preferentially lost during HCD before the peptide backbone fragments. EThcD preserves the glycan on the peptide during ETD fragmentation while the supplemental HCD generates oxonium ions for glycan identification, providing both peptide sequence and glycan information from the same spectrum.
What is the advantage of TMT labeling in glycoproteomics?
TMT multiplexing eliminates run-to-run variation in glycopeptide enrichment and LC separation, which are the dominant sources of technical variability in glycoproteomics. By labeling up to 16 samples with isobaric tags and combining them before enrichment, every glycopeptide experiences identical enrichment and chromatography across all samples. The TMT reporter ions in the low-mass region provide relative quantification with 8% average CV, as reported in systematic benchmarks.
Is site-specific glycosylation information possible from FFPE tissue?
Yes, with caveats. PNGase F digestion directly from FFPE sections can release N-glycans for glycomics analysis with acceptable reproducibility. For site-specific glycoproteomics from FFPE, intact glycopeptide analysis is more challenging due to formalin crosslinking reducing digestion efficiency. Specialized protocols using extended antigen retrieval, prolonged trypsin digestion, and higher input amounts can recover glycopeptides from FFPE, but coverage is substantially lower than from frozen tissue.
References:
- Deng Z, Wei R, Dong M, et al. Development of complementary enrichment strategies for analysis of N-linked intact glycopeptides and potential site-specific glycoforms in Alzheimer's disease. Talanta, 2026, 297:128595. doi:10.1016/j.talanta.2025.128595.
- Zhang H, Liao R, Donabedian P, et al. Improving Glycoproteomic Analysis Workflow by Systematic Evaluation of Glycopeptide Enrichment, Quantification, Mass Spectrometry Approach, and Data Analysis Strategies. Analytical Chemistry, 2024, 96(52):20481-20490. doi:10.1021/acs.analchem.4c04466.
- Hogan JD, Chalkley RJ, Krokhin OV. Comparative analysis of glycoproteomic software using a tailored glycan database. Analytical and Bioanalytical Chemistry, 2025, 417:2331-2342. doi:10.1007/s00216-025-05780-9.
- Sakaue K, Hanamatsu H, Nishikaze T, et al. Comparative Evaluation of Glycoproteomics Software for Rare Glycopeptide Identification. bioRxiv, 2025. doi:10.1101/2025.07.10.664079.
- Chalkley RJ, Baker PR, Medzihradszky KF, et al. Improved Glycopeptide Identification Using Protein Prospector. Molecular & Cellular Proteomics, 2025, 24(2):100903. doi:10.1016/j.mcpro.2025.100903.
- Chen W, Smeekens JM, Wu R. Comprehensive analysis of protein glycosylation by mass spectrometry. Proteomes. 2024;12(4):34. doi:10.3390/proteomes12040034.












