Global Drug Target Identification Service — From Phenotypic Hit to Confirmed Target
Six orthogonal chemoproteomics platforms. One integrated answer.
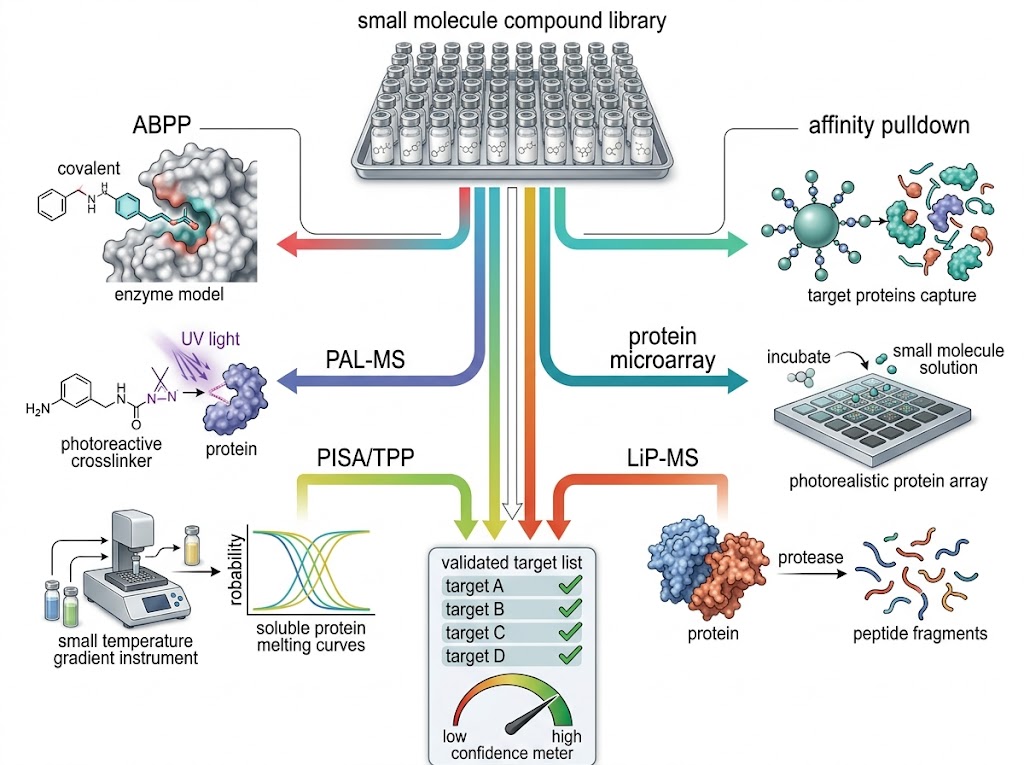
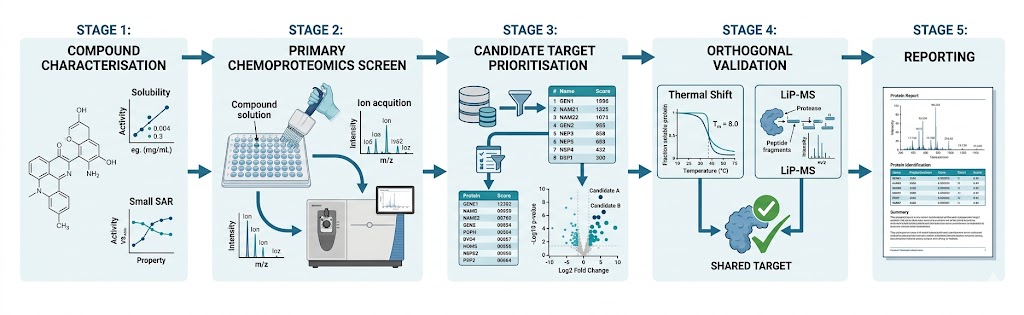
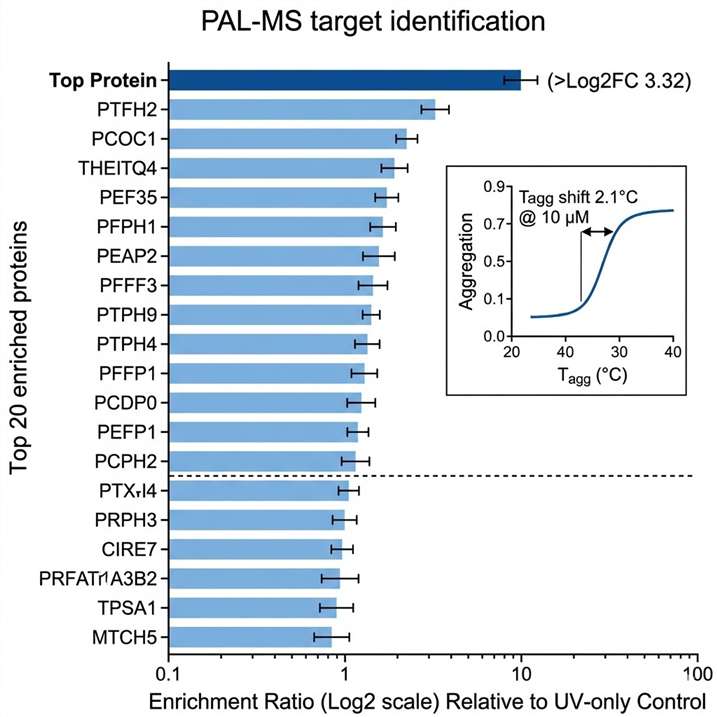
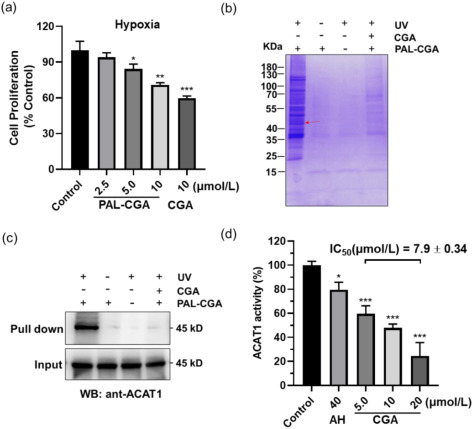
Every drug discovery program reaches a moment when the question shifts from "what does this compound do?" to "what protein does it bind?" Answering that question — definitively, with orthogonal evidence, and across diverse chemical matter — is the core mission of our Global Drug Target Identification service. We combine six orthogonal chemoproteomics and mass spectrometry platforms to map small molecule–protein interactions from phenotypic screening hits, natural products, covalent inhibitors, fragments, and approved drugs, delivering ranked target lists with cross-platform validation and statistical confidence.
At Creative Proteomics MassTarget, our target ID service is built for discovery teams who cannot afford false leads or single-method blind spots. Whether the starting point is a phenotypic hit from an HTS campaign, a bioactive natural product with an unknown mechanism, a covalent fragment series needing proteome-wide selectivity profiling, or a preclinical candidate requiring off-target de-risking, we design a multi-platform engagement matched to the physicochemical properties of your compound and the biological system in which it acts. For integrated multi-omics campaigns that combine target identification with broader pathway analysis, our multi-omics integration service provides a complementary framework for contextualising target findings within the wider cellular response.
Key Advantages:
- Six orthogonal chemoproteomics methods under one engagement — no single-method blind spots.
- Label-free and label-based options matched to compound type and biological context.
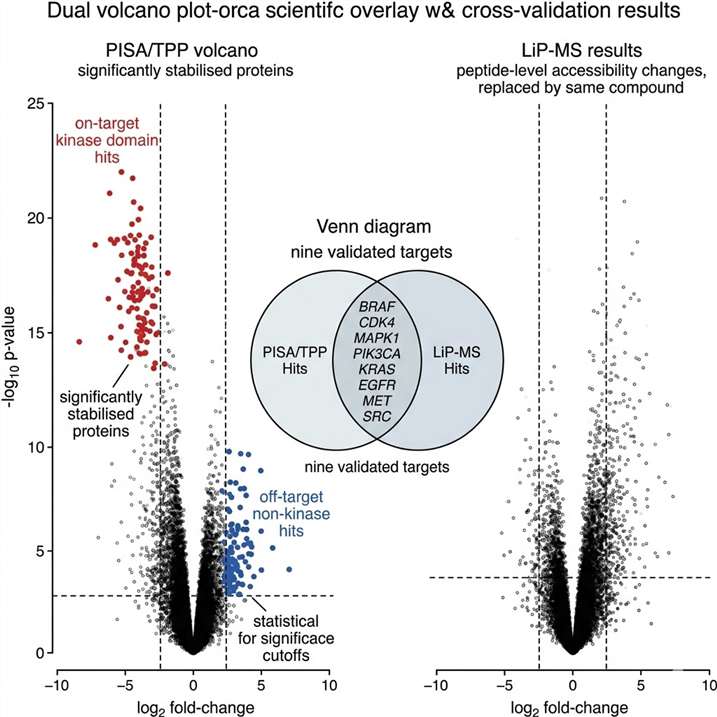
- Cross-platform target validation built into every project — targets reported only if confirmed by ≥2 independent methods.
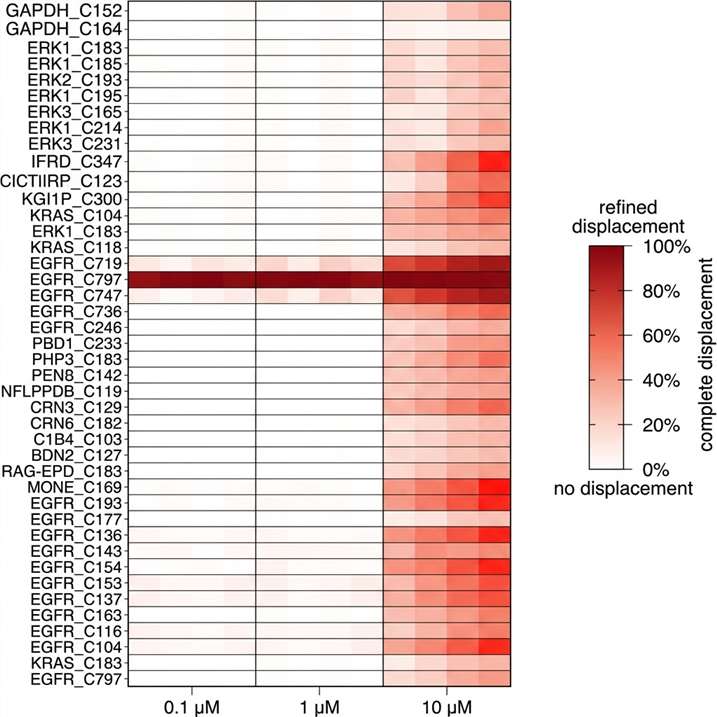
- Proteome-wide coverage: 5,000–9,000 proteins quantified per experiment depending on platform and cell type.
- Experience across diverse compound classes: covalent inhibitors, non-covalent binders, natural products, fragments, PROTACs, and clinical candidates.