Background
Identifying functional hits for challenging, unprecedented targets often suffers from high false-positive rates in conventional functional assays due to non-specific hydrophobic interactions or pan-assay interference compounds (PAINS). Researchers require a highly reliable, label-free, direct-binding confirmation approach to push their drug discovery pipelines forward and avoid wasting significant budget and time on synthesizing dead-end compounds.
Methods
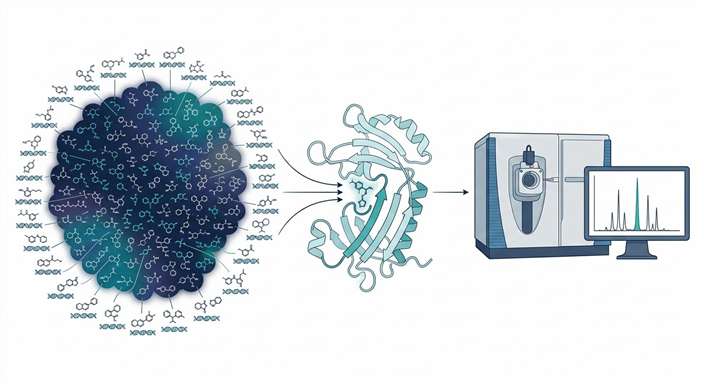
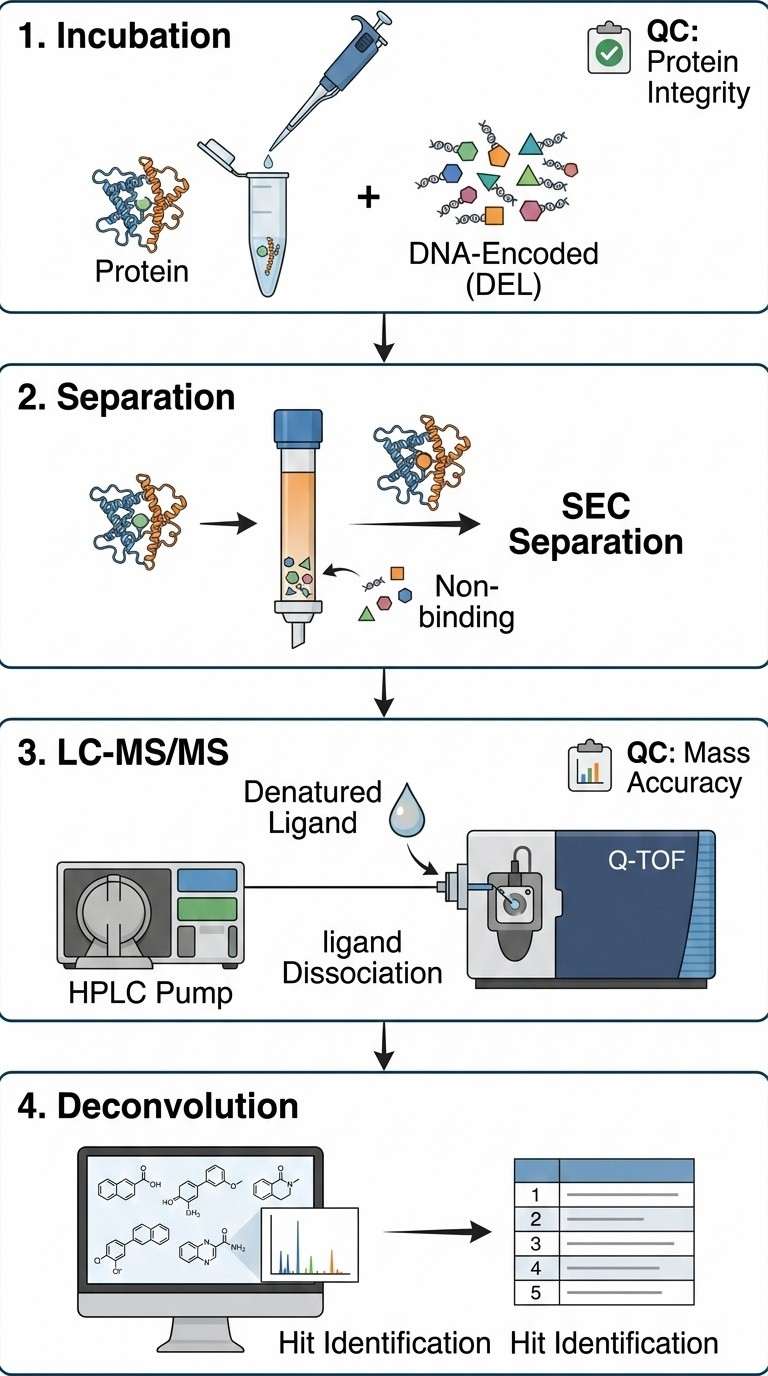
In a recent landmark study, researchers utilized an advanced iteration of ASMS known as Enantioselective Protein Affinity Selection Mass Spectrometry (E-ASMS methodology) to rapidly screen complex, highly diverse compound pools against target proteins. By utilizing chiral targets or ligands, this specific methodology provides an internal control for non-specific binding. The workflow incorporated robust SEC (Size Exclusion Chromatography) separation to clear the unbound library, followed by ultra-high-resolution MS detection to ensure absolute mass accuracy and differentiation of specific stereoisomers.
Results
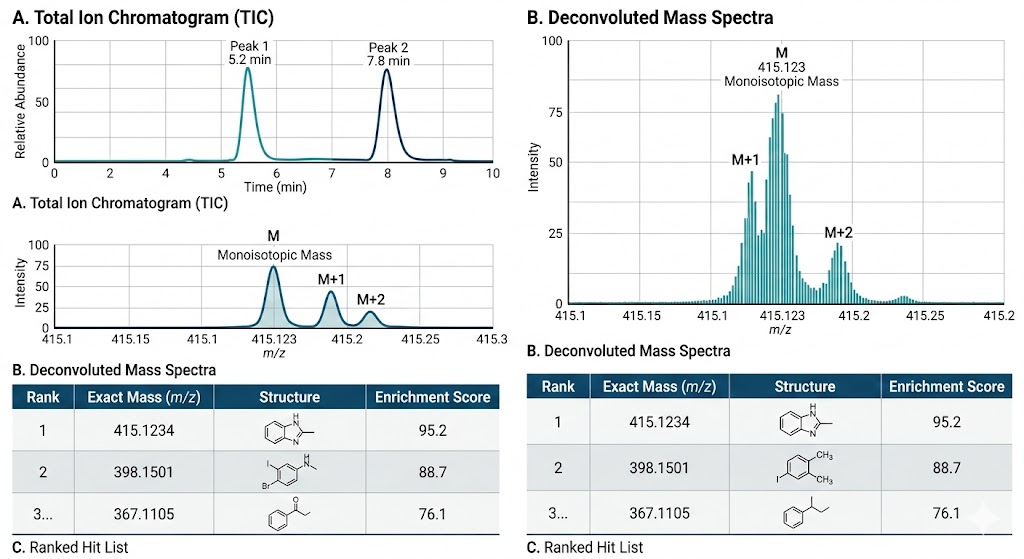
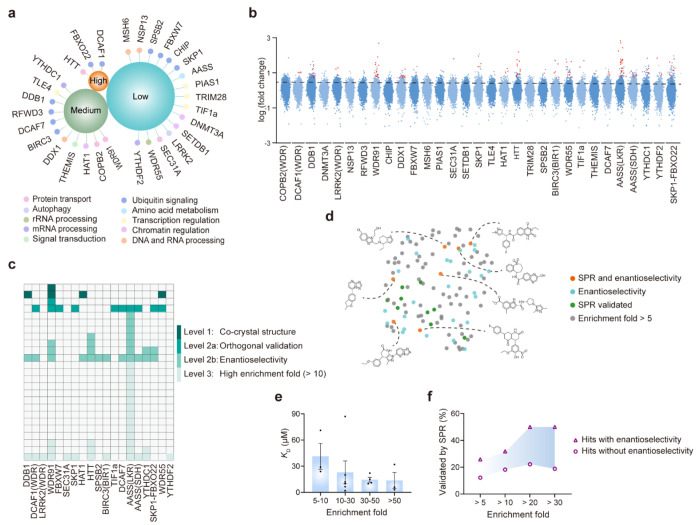
The advanced ASMS workflow successfully and definitively discriminated true high-affinity, stereospecific binders from background noise and non-specific matrix interactions. The E-ASMS method delivered a highly confident, pristine hit list that was validated by corresponding deconvoluted mass spectra, as clearly demonstrated in Figure 2 of the published research, which maps the robust signal enrichment of the true binders against the depleted background.
Conclusion
This pivotal research heavily highlights DEL-ASMS—and its advanced enantioselective variants—as an indispensable, gold-standard tool for accelerating hit discovery. It provides precise molecular weight evidence, confirms stereospecific interactions, and completely eliminates the assay interference that plagues complex, unstructured targets in modern drug discovery.