Background
Thermal proteome profiling has revolutionized label-free target discovery, allowing researchers to observe drug-protein interactions in their native cellular context. However, scaling the technology to the full proteome level relies heavily on robust data processing. As datasets grow larger and more complex, extracting true target engagement signals from the noisy background of a living cell requires highly optimized experimental designs and advanced statistical models.
Methods
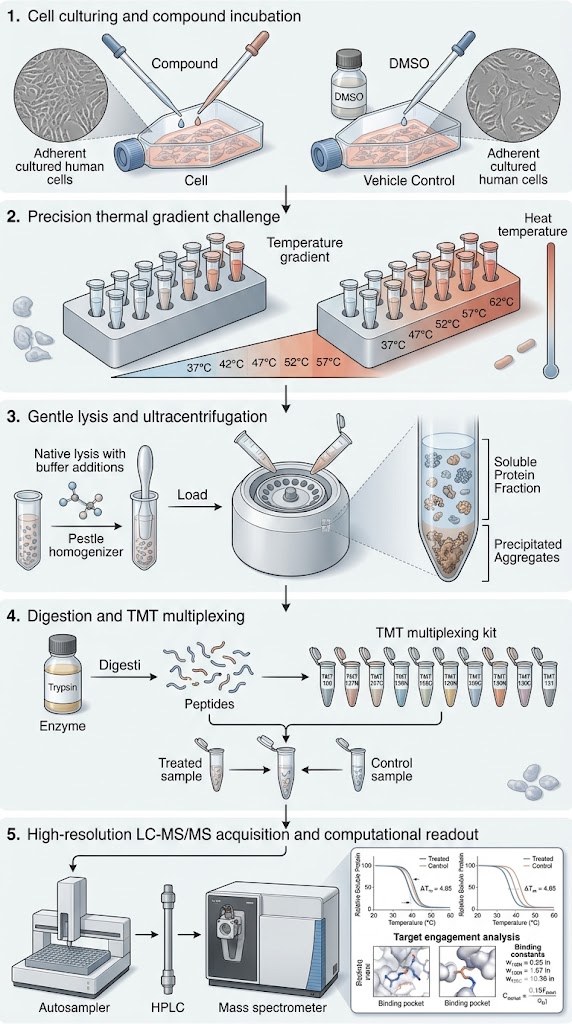
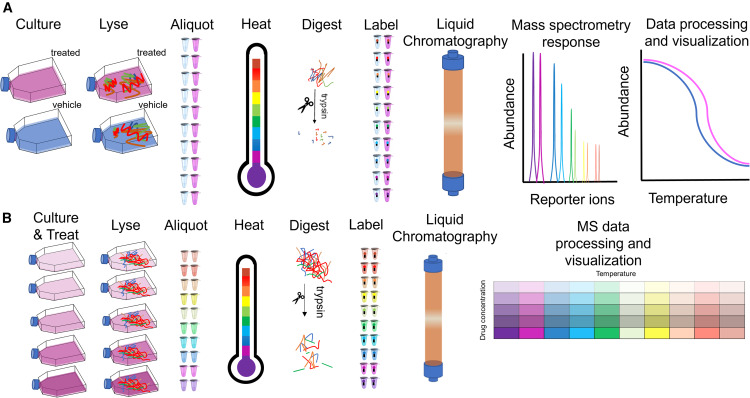
Researchers in a comprehensive study reviewed and implemented advanced experimental setups alongside sophisticated computational data analysis workflows for TPP. In the study's protocol, intact cells were treated with target compounds, subjected to precise thermal gradients, gently lysed, and analyzed via highly multiplexed mass spectrometry. The most crucial step of the methodology involved moving away from basic data processing and instead utilizing refined bioinformatics algorithms to mathematically fit melting curves and calculate statistical significance using non-parametric analysis of response curves (NPARC).
Results
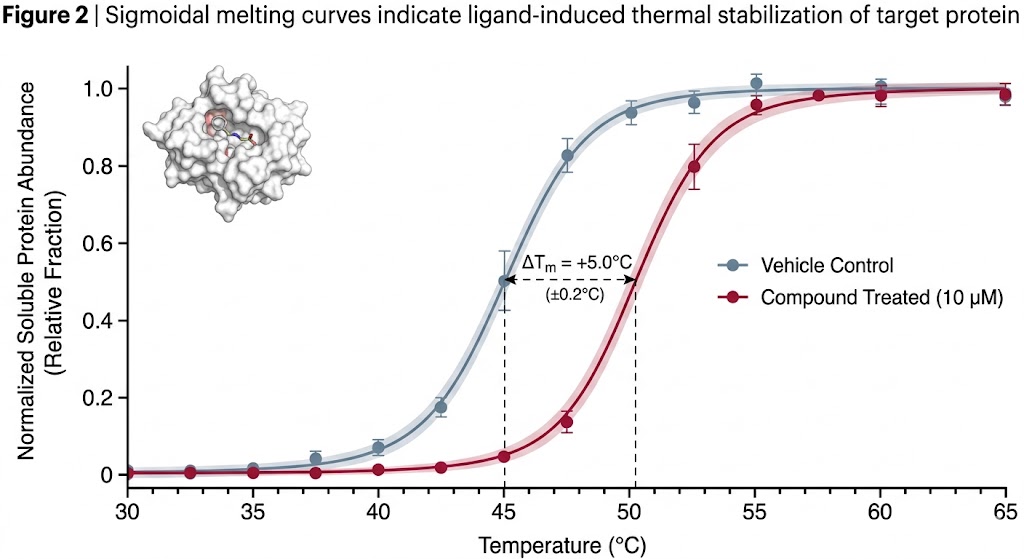
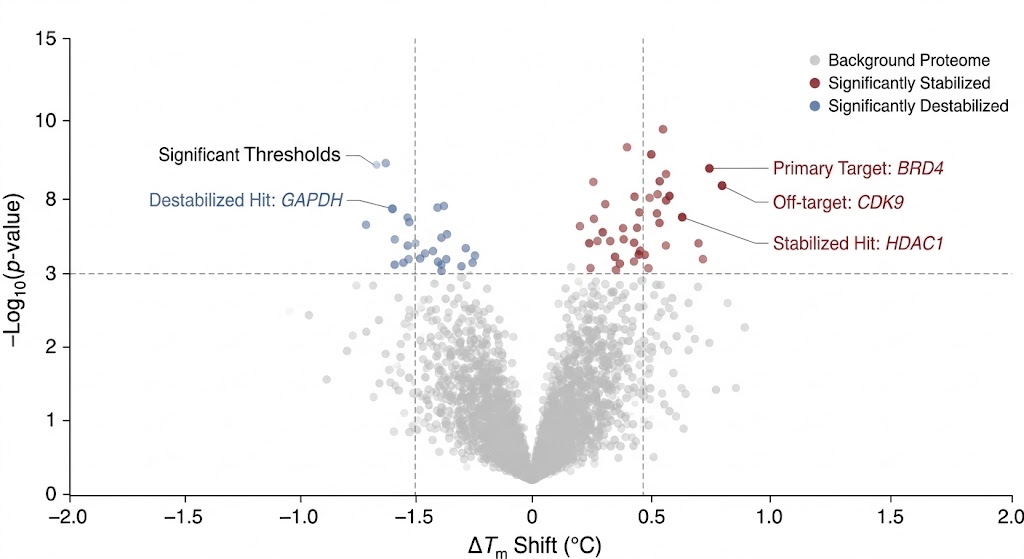
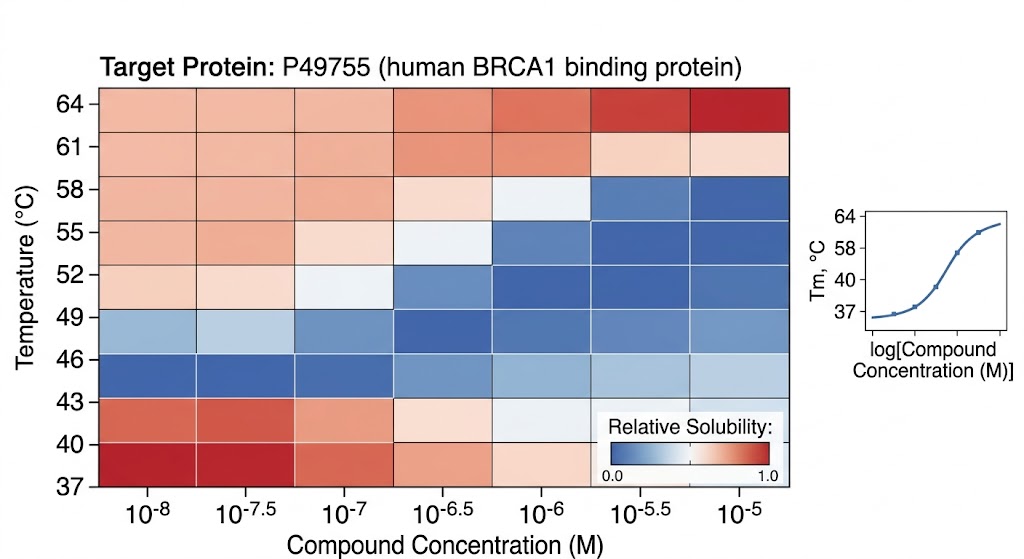
As clearly detailed and conceptualized in Figure 1 of the published research, the advanced computational workflow successfully transformed raw, noisy mass spectrometry abundance data into highly reliable, classic sigmoidal melting curves. The sophisticated algorithms effectively differentiated true drug-protein interactions from random thermal artifacts. This allowed the researchers to identify both primary therapeutic targets and novel off-target interactions with exceptionally high statistical confidence across the entire proteome.
Conclusion
This study proves that employing rigorous, state-of-the-art data analysis pipelines is mandatory for successful thermal shift proteomics. Advanced curve fitting and NPARC statistics ensure that researchers receive actionable, high-confidence target maps rather than noisy, uninterpretable data dumps, directly supporting confident lead optimization and compound de-risking.