Cross-Linking Mass Spectrometry Service for Protein Complex Structure and Interaction Analysis
Turn protein complex questions into structure-linked evidence with residue-pair proximity mapping, interface insight, and model-guided interpretation.
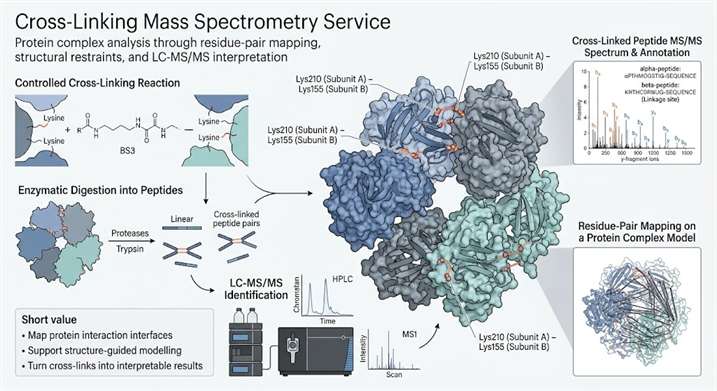
Cross-Linking Mass Spectrometry Service helps you turn protein complex questions into structure-linked evidence by capturing residue-pair proximity, supporting interface analysis, and adding experimental restraints for model-guided interpretation. Our XL-MS workflows are designed for protein interaction studies, complex assemblies, and structural biology projects that need more than simple interaction confirmation. For further reading on this topic, see our dedicated resource on amine-reactive crosslinker overview.
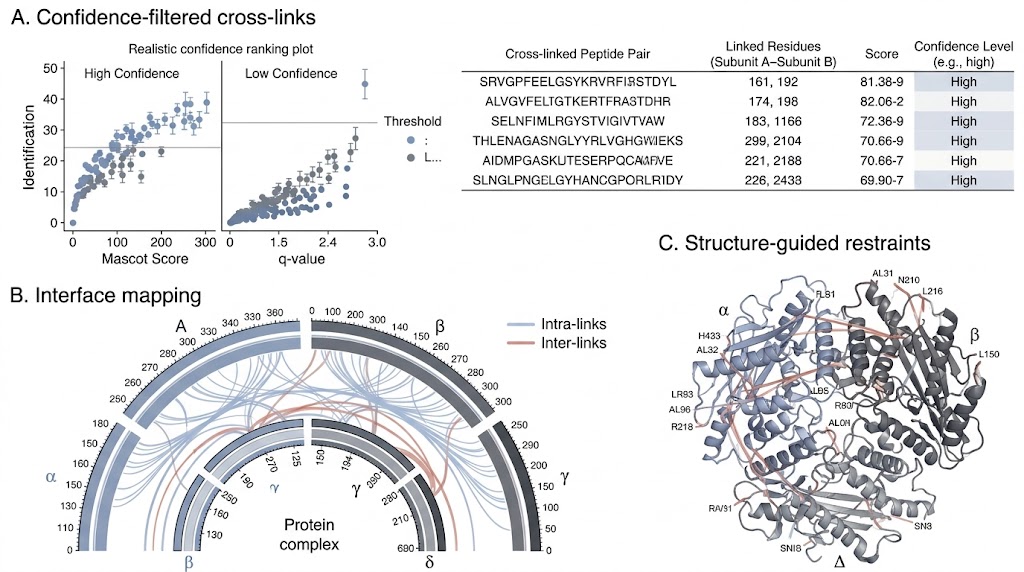
When you need more than a simple interaction yes-or-no answer, XL-MS can give you a different level of insight. It can help you understand which residues are close in space, which interfaces are supported by experimental evidence, and how structural restraints can strengthen protein complex analysis.
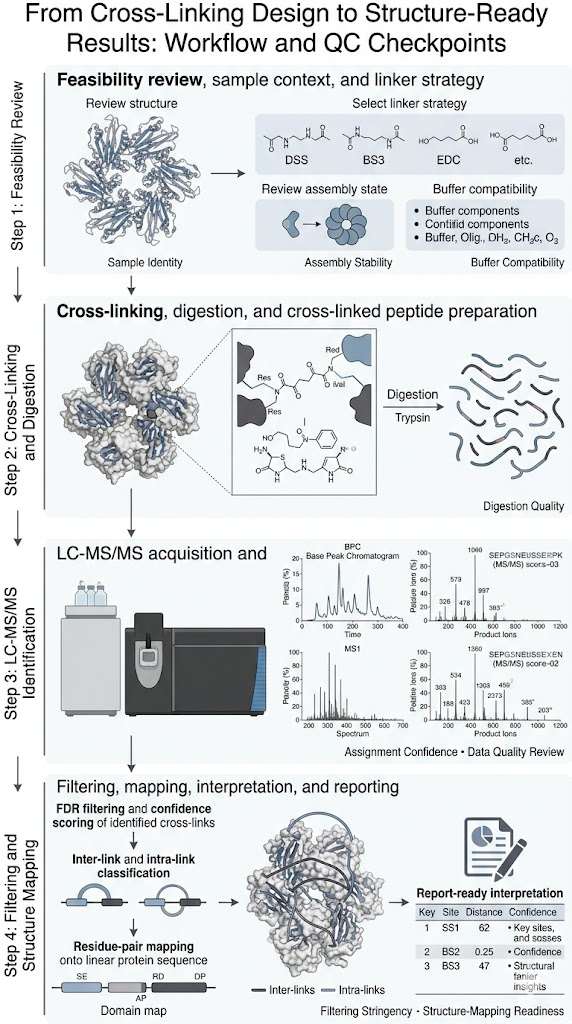
At Creative Proteomics, we approach XL-MS as a problem-solving service rather than a single experiment. That means we focus on project fit, linker strategy, sample feasibility, confidence-aware data analysis, and structure-ready outputs that help you move from raw identifications to interpretable structural decisions.
What we help you evaluate:
- Protein complex interfaces.
- Residue-pair proximity information.
- Structural restraints for modelling.
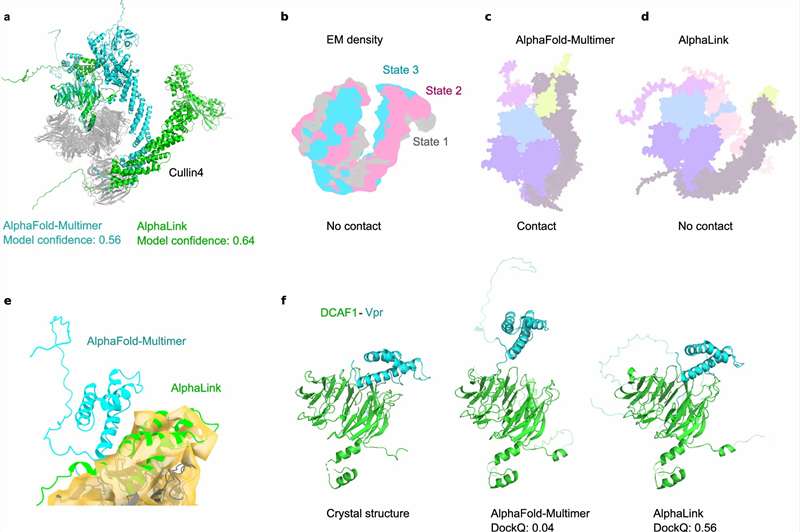
- Comparative state-dependent interaction changes.
- Structure-guided interpretation for complex systems.