- Service Details
- Case Study
Proteomics has been considered a comprehensive technique to decipher biological questions. The methodology regards to a range of separation methods including mass spectrometry(MS) The two principal approaches for protein identification and characterization via MS can be categorized as "bottom-up", which analyzing peptides by proteolytic digestion, and "top-down", namely global analysis of intact proteins by fragmentation.
What Is Top Down Proteomics?
The top down protein mass spectrometry strategy is a traditional proteomic analysis approach that involves breaking down large protein/enzyme complexes into smaller peptide fragments for analysis. Mass spectrometry techniques are widely applied in proteomics research.
Top-down proteomics analyzes intact proteins with high-throughput quality. Whereas, in bottom-up proteomics, proteins need to be digested into peptide fragments before MS analysis, Top-down proteomics involves separating intact proteins from complex biological samples using conventional separation techniques such as liquid chromatography or 2-D gel electrophoresis, followed by MS analysis. Both electrospray ionization (ESI) or matrix-assisted laser desorption/ionization (MALDI) considered as soft ionization method to generate ion molecules following with fragmentation via diverse dissociation methods, likely higher energy collision introduced dissociation (HCD), electron-capture dissociation (ECD) and electron-transfer dissociation (ETD), through tandem mass spectrometry, As a promising alternative strategy for protein identification, profiling, sequencing and PTM (post-translational modification) characterization, Top-down approach allows MS analysis of intact proteins that have not been cleaved, meaning the labile structural protein characteristics that are mostly destroyed in bottom-up MS are preserved. Hence, an universal characterization of modification sites and patterns can be achieved simultaneously in one spectrum with their supposed related correlations between these determined modifications. Meanwhile, protein digestion process elimination reduces time consumption during the experiment in contrast to bottom-up approach.
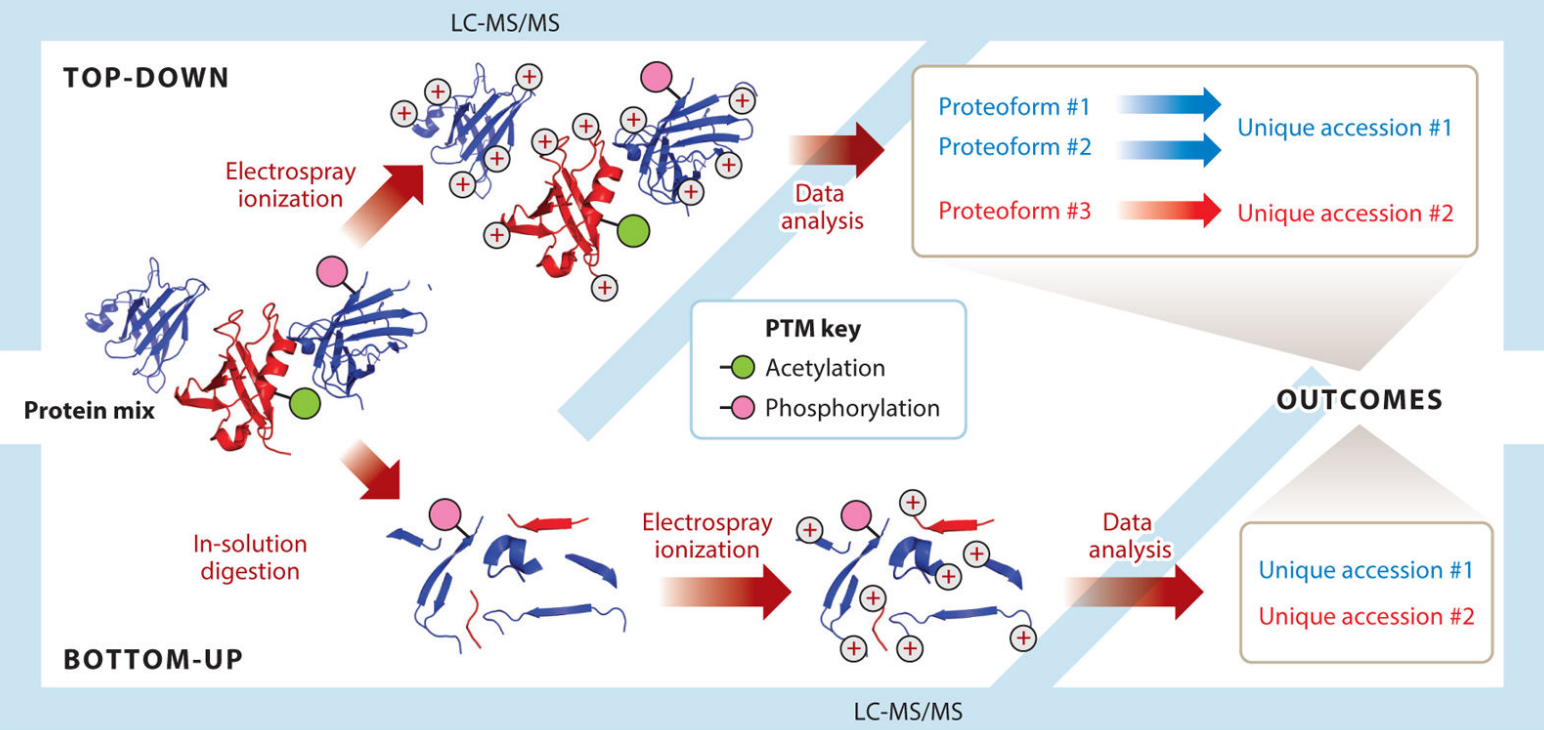 Figure 1. Divergent workflows in top-down and bottom-up proteomics. (Toby T K, et al.; 2016)
Figure 1. Divergent workflows in top-down and bottom-up proteomics. (Toby T K, et al.; 2016)
Top-down proteomics VS Bottom-up
Table 1. Comparison between bottom-up and top-down proteomics
| Bottom-up Proteomics | Top-down Proteomics | |
|---|---|---|
| Protein digestion | Yes | No |
| Detection of PTM | Yes | Yes |
| Analyzed sample forms | Peptide fragments | Full-length proteins |
| Detection efficiency | Relatively low | Significantly high |
| Strength | Well-developed for analyzing small peptides | Superior method for protein level sequence and PTM identification |
Top Down Proteomics at Creative Proteomics
Based on Creative Proteomics platform, we are capable of providing top-down proteomics services, including protein sequencing, PTMs characterization, and protein structure characterization.
Top-down MS maintains both single proteins analysis and protein complex characterization. Creative Proteomics supports both N- and C-terminal sequencing services under top-down strategy, particularly when the termini of your protein samples are blocked by modifications.
At Creative Proteomics, top-down mass spectrometry with ECD & ETD has been successfully applied to proteomics research, especially for sequence coverage mapping, identifying unpredicted PTMs identification, multiple modifications determination.
The protein structure characterization assist to a better understanding of protein fucntion. In addition, it plays an important role in biologic development and ongoing quality control. Currently, MS is commonly used to determine both the primary and higher order structures of proteins. For instances, the quaternary structure with disulfide bonds can be maintained and detected via top-down approach due to no digestion process has been applied under the native state of the protein sample.
Advantages of Top-down Proteomics
- Relatively simple sample preparation
- No protein digestion needed
- Direct detection of the molecular mass of biological proteins
- Mass information is retained in a great extent, such as PTMs
- Less time-consuming
Technical Platform
- Liquid Chromatography (LC)
- High Performance Liquid Chromatography (HPLC)
- Matrix Assisted Laser Desorption Ionization Mass Spectrometry (MALDI-MS)
Based on experienced staff and advanced instruments, Creative Proteomics provides a wide range of top-down proteomics with reasonable price in a time-saving manner. Our ordering procedure describles as follows. If you have any questions or specific requirements, please feel free to contact us.
Distinct hypertrophic cardiomyopathy genotypes result in convergent sarcomeric proteoform profiles revealed by top-down proteomics
Journal: Proc Natl Acad Sci U S A.
Published:2020
Abstract
Hypertrophic cardiomyopathy (HCM) is a common hereditary cardiac disorder and a significant contributor to sudden cardiac death among young individuals. While it is associated with mutations in genes responsible for encoding cardiac muscle proteins, the precise mechanism by which diverse mutations result in comparable clinical phenotypes remains elusive. To date, more than 1,400 gene mutations have been identified, adding to the intricate challenge of addressing this hereditary cardiac ailment.
Analysis Approach
1. In this investigation, the research group adeptly applied a top-down proteomics and phosphoproteome analysis approach grounded in high-resolution mass spectrometry. Their focus was directed towards dissecting surgical cardiac tissue specimens derived from hypertrophic cardiomyopathy patients. Through this meticulous endeavor, they illuminated a captivating phenomenon – a multitude of discrete gene mutations can orchestrate analogous modifications in cardiac muscle proteins. This study, thus, extends a comprehensive canvas for discerning the intricate protein attributes across both afflicted patients and their healthy counterparts.
Results
2. A Complex Sarcomeric Proteoform Landscape Revealed by Top-Down Proteomics.
 Fig. 1. A complex sarcomeric proteoform landscape.
Fig. 1. A complex sarcomeric proteoform landscape.
3. Phosphoproteome Analysis
3.1 Concerted Decrease in cTnI and ENH2 Phosphorylation in HCM.
 Fig. 2. Coordinated decrease in cTnI and ENH2 phosphorylation in HCM tissues
Fig. 2. Coordinated decrease in cTnI and ENH2 phosphorylation in HCM tissues
3.2 Altered Phosphorylation of cTnT, Tpm1.1, and MLC-2v in HCM.
 Fig. 3. Altered phosphorylation of cTnT, Tpm1.1, and MLC-2v.
Fig. 3. Altered phosphorylation of cTnT, Tpm1.1, and MLC-2v.
4. Top-down MS/MS Identification
Top-down MS characterization of cypher proteoforms in HCM.
 Fig. 4. Top-down MS characterization of cypher proteoforms in HCM.
Fig. 4. Top-down MS characterization of cypher proteoforms in HCM.
Conclusion
In this study, author used advanced top-down proteomics (LC-MS) to analyze cardiac sarcomeric proteoforms in HCM and donor heart tissues. We discovered complex proteoform patterns, including important Z-disk proteins like FHL2, ALP-H, and others. Using ultrahigh-resolution FTICR MS, we detailed the sequences of certain proteins.
Their analysis revealed distinct phosphorylation in sarcomeric proteins in HCM tissues compared to donor tissues. Interestingly, consistent proteoform alterations were observed in HCM patients' hearts, regardless of genetic mutations.
Notably, reduced phosphorylation of cTnI and ENH2 correlated strongly in HCM tissues, suggesting disrupted PKA signaling and potential interactions between myofilament and Z-disk proteins.
These findings highlight common pathways causing consistent proteoform changes in obstructive HCM, irrespective of genetic variations. This underscores the significance of proteoform-level understanding for phenotypic manifestations and potential treatments.
Future proteomics research in diverse HCM cases could offer valuable insights into disease progression and prognosis based on proteoform patterns. This may lead to novel therapeutic approaches targeting obstructive HCM broadly.
Reference
- Tucholski T, Cai W, Gregorich ZR, Bayne EF, Mitchell SD, McIlwain SJ, de Lange WJ, Wrobbel M, Karp H, Hite Z, Vikhorev PG, Marston SB, Lal S, Li A, Dos Remedios C, Kohmoto T, Hermsen J, Ralphe JC, Kamp TJ, Moss RL, Ge Y. Distinct hypertrophic cardiomyopathy genotypes result in convergent sarcomeric proteoform profiles revealed by top-down proteomics. Proc Natl Acad Sci U S A. 2020 Oct 6;117(40):24691-24700. doi: 10.1073/pnas.2006764117. Epub 2020 Sep 23. PMID: 32968017; PMCID: PMC7547245.













