- Service Details
- Case Study
What Is SRM/MRM
Selected reaction monitoring (SRM), also known as multiple reaction monitoring (MRM), is an important technique for quantitative of target proteins in complex sample. SRM/MRM technique can achieve rapid, sensitive, specific quantification of the target protein in a complex system and excellent quantitative reproducibility. SRM/MRM technology eliminates most of the non-target detection, that making the noise signal greatly reduced, and improves the detection sensitivity significantly. Compared to ELISA, SRM/MRM has better reproducibility, higher throughput, lower development cost and higher stability. At present, SRM/MRM has rapidly evolved into the representative technology of target technology, and is regarded as the "gold standard" for protein quantification based on mass spectrometry.
Real-time quantitative monitoring through MRM/SRM technology facilitates a range of studies including pharmacokinetics, clinical diagnostics, prohibited substance detection, industrial quality control for food and cosmetics, environmental monitoring, agricultural plant research, as well as metabolomics and research related to protein post-translational modifications and biomarker discovery based on proteomics.
SRM/MRM Principle
SRM/MRM technique is based on the known or assumed reactive ion information, targeted to select the data for mass spectrometry signal acquisition, removal of non-compliance with the interference of ion signals, through the data Mass spectrometry to obtain mass spectrometric quantitative information. The main instrument used in this technique is a triple quadrupole mass spectrometer, in which the first and third quadrupole are used as mass filters to specifically select peptide ions (precursor ions) that are identical to the pre-set mass to charge ratio. And the fragment ions (product ions) through the second quadrupole as the collision cell by CID (Collision Induced Dissociation) way to break the precursor ion to produce ions. SRM technology has a high selectivity, reduced the background noise, and interference of ions
Highlight of Mass Spectrometry MRM Technique
High Sensitivity: Through the use of two-stage ion selection, extensive interference ions are eliminated, reducing the chemical background of the mass spectrum. This significantly enhances the signal-to-noise ratio of the targeted analyte, achieving high sensitivity in detection.
Excellent Reproducibility: MRM selectively acquires mass spectral signals, avoiding the ionization of the target molecule, suppression of mass spectral signals, and the impact of in-source collision-induced fragmentation. As a result, reproducibility is correspondingly improved.
High Accuracy: Leveraging the specificity of MRM technology, consecutive enhanced ion scans are conducted, generating high-resolution tandem mass spectrometry (MS/MS) fragment data. Compared to full-scan and neutral loss mass spectrometry scan modes, this reduces the false positive rate of qualitative results during analysis, ensuring analytical accuracy.
High Throughput: Utilizing state-of-the-art mass spectrometry systems, MRM technology can handle as many as 300 pairs of precursor-fragment ions in each working cycle. This characteristic provides opportunities to investigate various modifications and abundance changes of numerous proteins, effectively meeting the research demands of proteomics.
No Need for Antibodies: Suitable for the validation and quantification of proteins for which quantitative analysis cannot be conducted using Western blotting or ELISA due to a lack of antibodies. This includes proteins from highly homologous protein families, post-translationally modified proteins (such as phosphorylated and methylated proteins), as well as proteins from plants, microbes, and model organisms.
Comparing SRM/MRM with Immunoassays
SRM/MRM differs from our commonly used label-free, iTRAQ/TMT methods for quantitative proteomics, both in terms of mass spectrometry hardware and data acquisition modes. SRM/MRM employs the selected scan mode based on triple quadrupole mass spectrometry. With the aid of this unique data acquisition approach, similar to WB and ELISA analyses, SRM/MRM allows for selective and specific quantitative (or absolute quantitative) analysis of target proteins.
So, since SRM/MRM functions similarly to WB and ELISA, what are the distinctions between the two?
SRM/MRM vs. WB, ELISA.
| Analysis Method | SRM/MRM | Immunoassay |
|---|---|---|
| Principle | Based on mass spectrometry, quantifying peptide segments for direct measurement of protein expression levels | Based on antibodies, quantifying protein-protein interactions indirectly to measure protein expression levels |
| High Specificity | Absolutely specific, capable of distinguishing PTMs, SNPs, variants, etc. | Awaiting evaluation (recently controversial) |
| High Throughput (Per Single Sample Analysis) | Over hundreds of targets | Up to 1 target (liquid chip technology can test dozens of targets, but the content is fixed) |
| Sensitivity | Moderate | High |
| Interference | None | ELISA: Self-antibodies, self-binding proteins |
SRM/MRM serves as a valuable companion to label-free, iTRAQ/TMT, and other omics technologies. Given the inevitable occurrence of quantitative false positives in large-scale omics techniques, results often necessitate validation through specific detection methods. The most conventional approaches include WB and ELISA, with SRM/MRM frequently employed for validation purposes as well.
SRM/MRM Analysis Services at Creative Proteomics
At Creative Proteomics, we provide specialized SRM/MRM analysis services tailored to your research needs. Our expertise in mass spectrometry and targeted proteomics ensures accurate and reliable results for your projects. With our advanced instrumentation and experienced team, we offer a comprehensive suite of SRM/MRM services, including method development, sample preparation, data acquisition, and data analysis. Our commitment to quality and precision makes us a trusted partner for your quantitative proteomics studies. Additionally, we also offer PRM (Parallel Reaction Monitoring) technology services that come with enhanced interference resistance and detection sensitivity.
SRM/MRM Assay workflow
- The SRM/MRM assay development. This is the core of the approach. First, 2 or 3 targeted peptides should be selected from a spectral library generation or Skyline software. The selected peptides should be unique to the protein of interest and easily detected by LC-MS. They also require no missed cleavage sites and no frequently modified amino acids. The product ions should also be selected. Second, detection conditions, such as fragmentation energy and cycle time, should be optimized. Third, a working linear curve was built with the corresponding concentration ratios on the x-axis and peak area ratios on the y-axis.
- Analysis of the sample. After digestion, the sample is performed on the triple quadrupole mass spectrometer.
- Data analysis. Data will be analyzed with Skyline software.
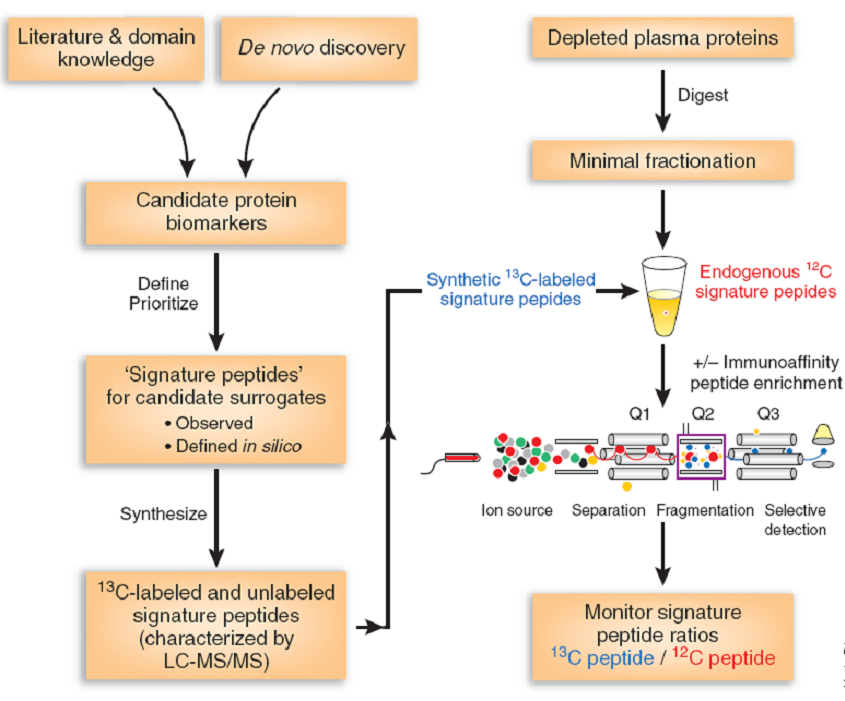
SRM/MRM application
- Verification of iTRAQ differential protein;
- Verification of label-free differential protein follow-up;
- Protein and peptide absolute quantification;
- Quantification of disease markers, the establishment of diagnostic models;
- Phosphorylation protein quantification, methylation protein quantification;
- Other post-translationally modified protein quantification;
- Quantitative analysis of pathways.
Sample Requirement
Protein Extracts: Concentration > 1 μg/μl, Total protein amount > 600 μg;
Cell Samples: Cell count > 10^7
Tissue Samples: Animal and microbial tissue wet weight > 10 mg
Plant tissue > 200 mg
Body Fluid Samples: Blood volume > 1 ml
Quantitation of putative colorectal cancer biomarker candidates in serum extracellular vesicles by targeted proteomics
Journal: Scientific Reports
Published: 06 October 2017
Abstract
Colorectal cancer (CRC) is one of the most prevalent malignant tumors in humans, ranking second in incidence among malignancies in developed Western countries. Research on biomarkers holds significant importance for the treatment of CRC. Many potential biomarkers for colorectal cancer have been revealed in numerous studies over the past few decades, yet there remains a lack of investigation into blood-based biomarkers. This article employs targeted proteomics as a means to conduct large-scale research on blood-based colorectal cancer targets, providing crucial insights for early diagnosis of CRC.
Research Method: Targeted Proteomics - SRM (Selected Reaction Monitoring)
Results
1. The authors conducted a search in Pubmed using the keywords "cancer + colorectal + expression" to identify potential biomarkers reported for colorectal cancer (CRC) between 2003 and 2014. These biomarkers were determined based on the following criteria: confirmation of differential protein expression in CRC through methods such as western blot and ELISA; relevance to CRC disease progression confirmed through RNA-related experiments; and potential biomarker identification through large-scale omics techniques. Due to the abundance of proteins in serum and the significant variability in protein abundance, comprehensive study of serum-based targets is challenging. Therefore, the authors focused on studying the extracellular vesicles (EVs) fraction within serum. They devised a strategy for identifying CRC targets: first employing shotgun proteomics to identify all proteins within EVs, then selecting potential CRC biomarkers from this pool. Subsequently, these potential targets were investigated using SRM for their precise quantification, ultimately confirming reliable and authentic CRC biomarkers.

2. Following the formulation of the identification strategy, the authors first extracted extracellular vesicles (EVs) from serum and CRC cells, respectively, and employed shotgun proteomics identification techniques to analyze all proteins within these two components. The identification results (Figure B - Venn diagram) indicated the types of proteins present in the serum and CRC cell EVs fractions, as well as the potential CRC biomarkers identified through literature searches, along with the numbers of overlapping intersections. The intersection between the proteins found in the serum and CRC EVs fractions and the potential CRC biomarkers from literature searches (highlighted in bold red font in the diagram below) represents the potential biological targets within the EVs fraction.

3. Subsequently, the authors employed targeted proteomics techniques utilizing Selected Reaction Monitoring (SRM) to identify the potential biomarkers that were previously confirmed (the identification process involved the selection of target peptides, SRM verification of target peptides, and targeted protein analysis). Serum samples were extracted from normal individuals, non-metastatic CRC patients, and metastatic CRC patients, and extracellular vesicles (EVs) were isolated from each of these samples for SRM identification. The figure below displays the content of selected target peptides in the three sample groups, providing insights into the expression levels of various biomarkers across these samples. Significance comparisons between the control group and the other groups are indicated in the figure (*p < 0.05, **p < 0.01, N.S: not statistically significant). The results revealed that the majority of target proteins belong to the Annexin protein family, including Annexin A3, A4, and A11.


4. In order to further investigate the sensitivity of the aforementioned biomarkers in early CRC detection, the authors utilized SRM to study the levels of CEA (a specific biomarker for late-stage metastatic CRC) in the serum samples of normal individuals, non-metastatic CRC patients, and metastatic CRC patients. The results indicated that the proportion of differential expression of CEA in non-metastatic CRC samples was only 38.8%, while in metastatic CRC samples, the proportion was 83.3%. This suggests that CEA functions as a more sensitive biomarker for late-stage CRC, with less prominent differences in early CRC identification. In comparison, the previously identified biomarkers in EVs exhibited sensitivities of at least 80% or more in both non-metastatic and metastatic CRC samples. This indicates that these target proteins can serve as biomarkers for both early and late-stage CRC, providing crucial evidence for early CRC detection.

5. Finally, in order to validate the reliability of the identified biomarkers, the authors selected a new set of patient samples and conducted identification of the previously identified target peptide segments within these samples. The results indicated consistency with the previous identification, providing further validation of the reliability of the aforementioned biomarkers.

References
- Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nature Methods. 2012; 9(6): 555-66.
- Addona TA, Shi X, et al. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nature Biotechnol. 2011; 29(7): 635-43.
- Shiromizu, T., Kume, H., Ishida, M. et al. Quantitation of putative colorectal cancer biomarker candidates in serum extracellular vesicles by targeted proteomics. Sci Rep 7, 12782 (2017).











