Tandem Affinity Purification (TAP)-MS Service
Map native protein complexes with confidence—TAP-MS at Creative Proteomics isolates your bait under gentle, orthogonal purifications and identifies bona fide interactors with high-resolution LC-MS/MS and rigorous statistics.
- Cut through background: Two-step affinity capture to suppress non-specific binders
- Keep complexes intact: Mild buffers and on-bead/TEV elution to preserve weak interactions
- Quantify changes: LFQ, SILAC, or TMT multiplexing for condition-dependent interactomes
- Decide fast: Ranked interactors, stoichiometry estimates, and publication-ready figures
- All matrices: Mammalian, yeast, bacterial systems, tissues, organoids, and cell-free preps
Submit Your Request Now
×
What You Will Receive
- Raw LC-MS/MS data (vendor format + mzML)
- Identified proteins with FDR control
- Quantitative interactor list with fold-change and q-values
- Final report (Excel + PDF)
- When to Choose
- Advantages
- Workflow
- Methods
- Sample Requirement
- Data Analysis
- FAQs
What Is TAP-MS and Why It Matters
Tandem Affinity Purification–Mass Spectrometry (TAP-MS) combines sequential, orthogonal affinity steps (e.g., Protein A–TEV–CBP or Strep–FLAG) with high-resolution LC-MS/MS to isolate a tagged "bait" protein together with its associated partners. Compared with single-step IP-MS, TAP-MS delivers higher purity and higher confidence in interaction calls, particularly for low-abundance, transient, or membrane-associated complexes.
Common hurdles it solves
- High background from sticky proteins or abundant chaperones → orthogonal elutions dramatically reduce carry-over.
- Antibody variability in classical IP → tag-based capture with standardized resins.
- Loss of weak/transient interactors under harsh wash conditions → gentle buffers and optional crosslinking preserve biologically relevant contacts.
- Ambiguous "hit" lists without statistics → model-based scoring (e.g., SAINT/MiST), CRAPome filtering, and FDR control separate signal from noise.
Principle of TAP-MS
Express a dual-tag bait → perform two orthogonal captures → acquire LC-MS/MS data for both experimental and matched control samples → subtract control-enriched proteins → apply statistical models to call high-confidence interactors.
When to Choose TAP-MS over Single-Step IP-MS
You should consider TAP-MS if:
Background noise is a concern
→ You're working with lysates prone to non-specific binding (e.g., nuclear, membrane-rich, or microbial extracts), and need to minimize false positives through orthogonal enrichment.
Your target is part of a weak or transient complex
→ TAP-MS uses mild elution conditions and optional crosslinking to retain labile interactions often lost in single-step IP.
You require high-confidence interaction data for publication or pathway modeling
→ TAP-MS combines two-step purification with statistical scoring (SAINT/MiST) and control subtraction, delivering clean, ranked interactors ready for interpretation.
Antibody availability or quality is limiting
→ TAP-MS leverages standardized epitope tags and validated resins, eliminating dependency on variable antibody performance.
Your study involves comparative interactomics
→ Whether across treatments, mutations, or time points, TAP-MS supports TMT, SILAC, or label-free quantification with high reproducibility.
In short, choose TAP-MS when your research demands clarity over complexity, especially in systems where traditional IP is insufficiently selective or where biological conclusions depend on interaction specificity.
Why Choose Creative Proteomics for TAP-MS
- Specificity boost: Orthogonal two-step enrichment typically reduces non-specific background ≥10× versus single-step IP under matched buffers, improving positive-predictive value for interaction calls.
- Depth & dynamic range: Orbitrap/timsTOF platforms capture >10⁵ dynamic range with ≤3 ppm mass accuracy for confident peptide-spectrum assignments.
- Reproducibility: Median replicate CVs typically ≤10% (TMT) and ≤15% (LFQ) at the protein level for TAP-enriched samples.
- Quant you can trust: SPS-MS3 on Orbitrap Fusion-class instruments minimizes reporter interference, improving ratio fidelity for multi-condition interactomes.
- Actionable stoichiometry: iBAQ-based relative abundance enables rank-order stoichiometry and complex composition assessment with high replicate correlation.
Workflow for TAP-MS Analysis
1. Study Design — Define bait/tag architecture, biological comparisons, controls (empty tag/unrelated bait), and quant scheme (LFQ/TMT/SILAC).
2. Sample Intake & QC — Expression/enrichment check (if provided), protein load assessment, buffer compatibility verification.
3. First Capture — Gentle, MS-compatible buffers; IgG/Strep/anti-FLAG resin capture; matrix-tuned washes.
4. Orthogonal Elution & Second Capture — TEV cleavage or desthiobiotin elution, followed by Calmodulin/FLAG/Strep re-capture.
5. Proteolysis & Cleanup — On-bead or in-solution trypsin/Lys-C; desalting; optional high-pH fractionation for depth.
6. LC-MS/MS — Instrument/mode matched to goals (DDA/DIA/SPS-MS3).
7. Computation & Statistics — Identification, FDR, quant modeling, background correction, interactor scoring, pathway/network analysis.
8. Reporting — Ranked interactors with q-values, fold-changes/ratios, stoichiometry estimates, QC dashboards, figures, and all raw/processed files.

Methods & Instrumentation Used for TAP-MS Analysis
Thermo Orbitrap Fusion Lumos
- MS1 resolution: 120,000 @ m/z 200
- MS2 (HCD) resolution: 30,000–60,000
- Supports SPS-MS3 for accurate TMT quantification
- Mass accuracy: ≤3 ppm
- Ideal for: TMT-based comparative interactomics, deep coverage of complex assemblies
Thermo Q Exactive HF-X
- MS1 resolution: up to 120,000
- High-speed TopN DDA acquisition
- Ideal for: label-free interactome profiling, high-throughput screening
Thermo EASY-nLC 1200 (nano-flow LC system)
- C18 reverse-phase columns: 75 µm × 25–50 cm, 1.6–1.9 µm particles
- Optimized for low sample input and complex peptide separation

Orbitrap Fusion Lumos (Figure from Thermo)

Q Exactive HF-X MS (Figure from Thermo)

EASY-nLC 1200 (Figure from Thermo)
Sample Requirements for TAP-MS Analysis
| Matrix / Source | Recommended Tags | Minimum Input (per replicate) | Lysis & Buffers (MS-compatible) | Additives & Notes | Shipping |
|---|---|---|---|---|---|
| Mammalian cells (adherent/suspension) | TAP (ProtA–TEV–CBP), Twin-Strep, 3×FLAG, HA/FLAG, His/FLAG | ≥2–5 mg total protein (≈ 1–5 × 10⁷ cells) | Non-denaturing (0.1–0.5% NP-40/Triton; moderate salt); avoid SDS/high glycerol | Protease/Phosphatase inhibitors; DNase optional; provide vector map and expected MW | Pellets flash-frozen; dry ice |
| Yeast | TAP, 3×FLAG, Twin-Strep | ≥2–5 mg total protein | Bead-beating or cryo-pulverization; mild detergent | Include auxotrophic marker info and growth conditions | Pellets frozen; dry ice |
| Bacteria | Twin-Strep, Strep-FLAG, His/FLAG | ≥2–5 mg total protein | Lysozyme/sonication under mild, salt-balanced conditions | Reduce endogenous proteases; clarify lysate thoroughly | Pellets frozen; dry ice |
| Tissues/Organoids | Strep/FLAG; pre-validated TAP | ≥50–200 mg wet tissue | Dounce/bead-mill; non-denaturing | Minimize lipids/particulates; include matrix-matched controls if possible | Tissue powder/pieces; dry ice |
| Alternative Inputs (purified protein / gel bands) | Any dual-tag or native complex | Purified protein: ≥ 5–10 µg at ≥ 0.1 µg/µL; SDS-PAGE bands: clearly stained, excised single band | Exchange purified complexes into low-salt, detergent-free buffer prior to shipment | Best for targeted follow-ups; interaction partners reported only if complex is retained | Cold packs or dry ice per buffer; consult first |
Demo Results
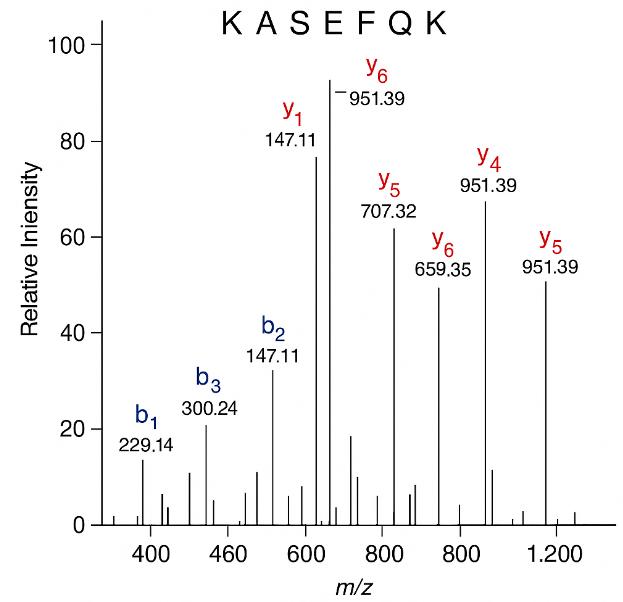
Representative annotated MS/MS spectrum of peptide KASEFQK, showing matched b- and y-ion fragments supporting confident identification.
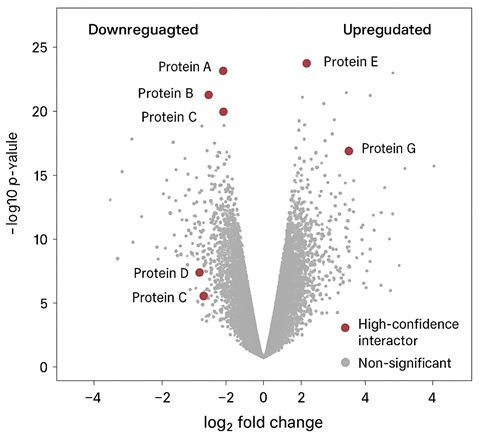
Volcano plot of TAP-MS results showing statistically significant interactors enriched relative to control. Red points indicate high-confidence interactors with |log₂FC| > threshold and p < 0.05.
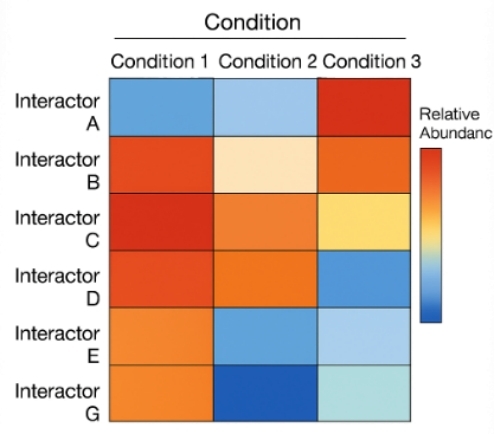
Heatmap showing relative abundance of interactors across three experimental conditions, quantified by TMT or LFQ. Color intensity reflects normalized signal levels.
Use Cases for TAP-MS
High-Confidence Mapping of Protein Complexes
TAP-MS isolates native complexes with minimal background, ideal for resolving core subunits and modular assemblies.
Detection of Weak or Transient Interactions
Mild elution and optional crosslinking preserve labile partners often lost in single-step IP, enabling detection of low-affinity interactors.
Comparative Interactomics Across Conditions
Supports TMT, SILAC, or LFQ-based quantification to reveal interaction changes across treatments, genotypes, or time points.
Target Validation Under Perturbation
Identify context-specific interactors in response to drug treatment, pathway activation, or gene knockdown.
Functional Annotation of Uncharacterized Proteins
Use TAP-tagged ORFs to infer function through co-purifying partners, accelerating pathway mapping.
Dissecting Domain-Specific Binding
Apply truncated or domain-mutant baits to define interaction interfaces and modular interaction logic.
FAQ of TAP-MS Assay
How do I choose a tag architecture that preserves bait functionality?
Select tag orientation (N- or C-terminal) based on domain structure and known functional sites. Include flexible linkers to minimize steric interference. For uncharacterized proteins, validate both tag locations with localization and expression tests before scaling.
Is TAP-MS suitable for membrane-bound or chromatin-associated protein complexes?
Yes. Use MS-compatible mild detergents (e.g., digitonin, DDM) for membrane targets and salt-tuned, benzonase-containing buffers for chromatin complexes. Adjust wash stringency to reduce background while preserving native interactions.
When should crosslinking be applied in TAP-MS workflows?
Use crosslinking selectively when targeting weak or transient complexes. Choose MS-cleavable or reversible crosslinkers and validate their impact on elution efficiency and downstream analysis. Avoid over-crosslinking that may reduce peptide recovery.
What control samples are necessary to ensure interactor specificity?
Include an empty-tag control processed identically to the bait. Unrelated baits or resin-only samples can further identify tag- or resin-specific binders. Controls must undergo both purification steps for valid subtraction and scoring.
How many biological replicates are recommended for TAP-MS?
A minimum of three biological replicates per condition is recommended to support statistical interactor scoring (e.g., SAINT, MiST) and ensure robust FDR estimation. Prioritize independent cultures or preparations over technical repeats.
Can I use TAP-MS with transiently expressed or weakly expressed baits?
Yes, but low expression may require scaling up cell input. Use optimized lysis, gentle handling, and if needed, high-sensitivity LC-MS modes. Endogenous tagging or codon optimization may also improve recovery.
Can TAP-MS be combined with TMT, SILAC, or label-free quantification?
Absolutely. TMT enables multiplexed comparison with high precision (use SPS-MS3 for accuracy), SILAC allows accurate pairwise contrasts, and label-free methods are ideal when metabolic labeling is not feasible.
How should TMT channels be allocated for bait and control samples?
Distribute bait and control samples across the TMT plex to avoid ratio distortion. Include replicates and use a pooled reference channel if comparing multiple conditions across batches.
What quality control metrics indicate a successful TAP-MS experiment?
Key QC indicators include high bait coverage, low background (compared to CRAPome), replicate correlation, and consistent peptide-level quantification. Visual dashboards in reporting help confirm pull-down integrity.
How does TAP-MS statistically distinguish true interactors from contaminants?
Statistical tools like SAINT or MiST score proteins based on control subtraction and enrichment magnitude. Combined with CRAPome frequency filtering and q-value thresholds, this ensures confident interactor calls.
Can TAP-MS help identify RNA- or DNA-mediated interactions?
Yes. Conduct parallel purifications ±RNase or DNase to assess nucleic acid dependency. A sharp decrease in interactor signal post-nuclease suggests RNA/DNA-mediated association.
What is the difference between on-bead digestion and elution-based digestion?
On-bead digestion simplifies processing but may introduce resin background. Elution followed by in-solution digestion increases cleanliness and allows for deeper peptide fractionation.
When is high-pH fractionation recommended?
Use peptide-level fractionation when deep coverage or stoichiometric ranking is required, especially for complex interactomes. Consider trade-offs between depth and potential dilution of low-abundance peptides.
How can I minimize free biotin interference in Strep-tag workflows?
Avoid biotin-supplemented media, clarify lysates thoroughly, and include a pre-wash step before elution. Monitor carry-over in controls to ensure specificity.
What should I do if my bait recovery is poor?
Check expression levels, optimize lysis conditions, and ensure the resin-tag pair is compatible. You may also try reversing the tag orientation or using an alternative capture resin for improved yield.
Can TAP-MS provide stoichiometric insights into protein complexes?
Yes. Use iBAQ or peptide-level intensity data to estimate relative stoichiometry within complexes. While semi-quantitative, these values can guide rank-based interpretation of complex composition.
How can I preserve labile post-translational modifications in TAP-MS?
Include inhibitors (e.g., phosphatase, deubiquitinase) in lysis and wash buffers, avoid excessive detergent or salt, and process samples rapidly. Be cautious with additives that may suppress ionization during MS.
Is endogenous tagging compatible with TAP-MS?
Yes. CRISPR/Cas9-mediated tagging supports native expression levels and improves biological relevance. Ensure that knock-in lines are validated for expression, localization, and functional activity.
What database and search parameters should I provide for non-model species?
Submit a custom FASTA file including all isoforms, tag sequences, and common contaminants. Document search settings, decoy strategies, and modifications for transparent data interpretation.
How do I minimize batch effects in multi-sample TAP-MS experiments?
Randomize sample processing and injection order, include a pooled reference, and normalize using internal standards or bridge channels when using TMT.
Learn about other Q&A about proteomics technology.
Publications
Here are some publications in Proteomics research from our clients:

- Extracellular vesicles from the human natural killer cell line NK3. 2021. https://doi.org/10.3389/fcell.2021.698639
- Trypanosoma cruzi DNA Polymerase β Is Phosphorylated In Vivo and In Vitro by Protein Kinase C (PKC) and Casein Kinase 2 (CK2). 2022. https://doi.org/10.3390/cells11223693
- Synergistic Combined-proteomics Guided Mapping strategy identifies mTOR mediated phosphorylation of LARP1 in nutrient responsiveness and dilated cardiomyopathy. 2022. https://doi.org/10.1101/2022.10.13.512080
- The cytoplasmic microtubule array in Neurospora crassa depends on microtubule-organizing centers at spindle pole bodies and microtubule+ end-depending pseudo-MTOCs at septa. 2022. https://doi.org/10.1016/j.fgb.2022.103729
- Hyperactivation of the proteasome core particle in Caenorhabditis elegans protects against proteotoxic stress and extends lifespan. 2021. https://doi.org/10.21203/rs.3.rs-960676/v1






