Subcellular Proteomics Service for Accurate Protein Localization and Drug Targeting
Subcellular proteomics—combining organelle isolation with high-resolution LC-MS/MS—enables precise mapping of proteins within cellular compartments, revealing their localization, movement, and mis-regulation, without the ambiguity of whole-cell digests.
our workflow isolates individual organelles and surface membranes so you see exactly which proteins act, move, or mis-regulate in each compartment. By combining ultra-clean fractionation with high-resolution LC-MS/MS, we routinely detect >1,100 compartment-specific proteins—even those present at femtomole levels—helping you:
- Detect low-copy biomarkers hidden in bulk proteomes
- De-risk target discovery by confirming spatial co-localization with drug-pathway nodes
- Track dynamic relocalization under treatment or stress to reveal actionable mechanisms
Submit Your Request Now
×
- Picogram-level sensitivity – Orbitrap Fusion Lumos & timsTOF Ultra 2 quantify proteins from as little as 25 pg digest
- >40 % deeper coverage vs. whole-cell digests thanks to proprietary density-gradient and affinity purification steps
- Label-free DIA, TMT/iTRAQ, or SILAC quantitation in a single platform for flexible experimental design
- AI-assisted localization & interaction mapping using MaxQuant, Spectronaut and dynamic organellar-map algorithms
- Publication-ready deliverables: raw data, QC metrics, volcano plots, interaction networks, and expert review session.
- What is
- Service Details
- Case Study
- FAQ
- Publication
Subcellular Proteomics: Mapping Cellular Machinery for Deeper Insights
Modern drug discovery and disease research demand precise knowledge of where proteins operate inside cells. That's the essence of subcellular proteomics service—exploring the proteome not as a single mass but as an organized map of cellular compartments. This approach gives researchers the context needed to interpret proteomic analysis accurately and pinpoint protein function.
Unlike whole-cell proteomics, which can involve over 12,000 to 40,000 proteins, subcellular proteomics narrows the focus to smaller compartments, each typically containing between 500 and 4,000 proteins.
This simplification improves detection of low-abundance proteins, which are often crucial for identifying biomarkers or therapeutic targets.
Researchers have long relied on biochemical fractionation techniques—using density, size, and charge differences—to isolate subcellular structures and minimize contamination. These foundational methods now empower more refined proteomic workflows.
How Subcellular Proteomics Drives Discovery
Advances in LC-MS/MS technology have elevated subcellular proteomics from theory to powerful practice. Prefractionation paired with high-resolution mass spectrometry allows scientists to dissect complex protein mixtures and gain unprecedented detail about cell biology.
1. Recent case studies highlight how targeted compartment analysis leads to:
2. Improved sensitivity for detecting low-copy proteins.
3. Better functional annotation of proteins tied to diseases or therapeutic pathways.
4. Deeper understanding of cellular dynamics under different physiological conditions.
Creative Proteomics brings together cutting-edge instrumentation and expert bioinformatics to deliver a comprehensive subcellular proteomics service tailored for industry and research.
Our Subcellular Proteomics Service at Creative Proteomics
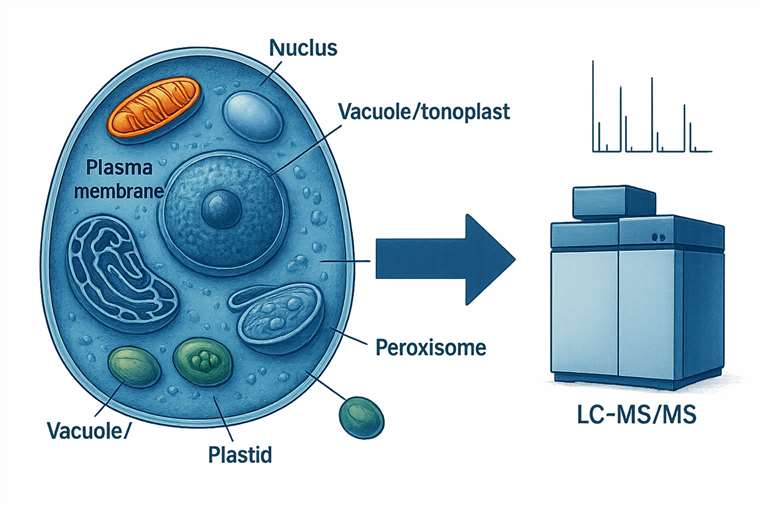
At Creative Proteomics, we leverage Thermo Fisher's Orbitrap Fusion Lumos mass spectrometer and nanoLC-MS/MS to push the boundaries of subcellular proteomic analysis. Our experience covers multiple cellular structures, enabling qualitative and quantitative studies of critical compartments such as:
Biological membranes govern key life processes through their resident proteins. Notably, around half of all known drugs target membrane proteins. Studying membrane proteomes helps identify disease biomarkers and discover novel drug targets.
Chloroplast Proteomics
Chloroplasts, essential for photosynthesis and containing their own genetic material, hold clues to plant growth, development, and metabolism. Chloroplast proteomics supports research into crop optimization and plant-based therapeutics.
Mitochondrial Proteomics
Often called the "powerhouses of the cell," mitochondria regulate energy metabolism and cell survival. Altered mitochondrial proteins can trigger metabolic disorders or degenerative diseases. Mitochondrial proteomic analysis provides insights crucial for developing mitochondria-targeted therapies.
Exosomes—tiny vesicles involved in cell communication—play roles in tumor progression, immune responses, and tissue repair. Exosome proteomics reveals molecular cargo linked to disease mechanisms and potential clinical applications.
Workflow
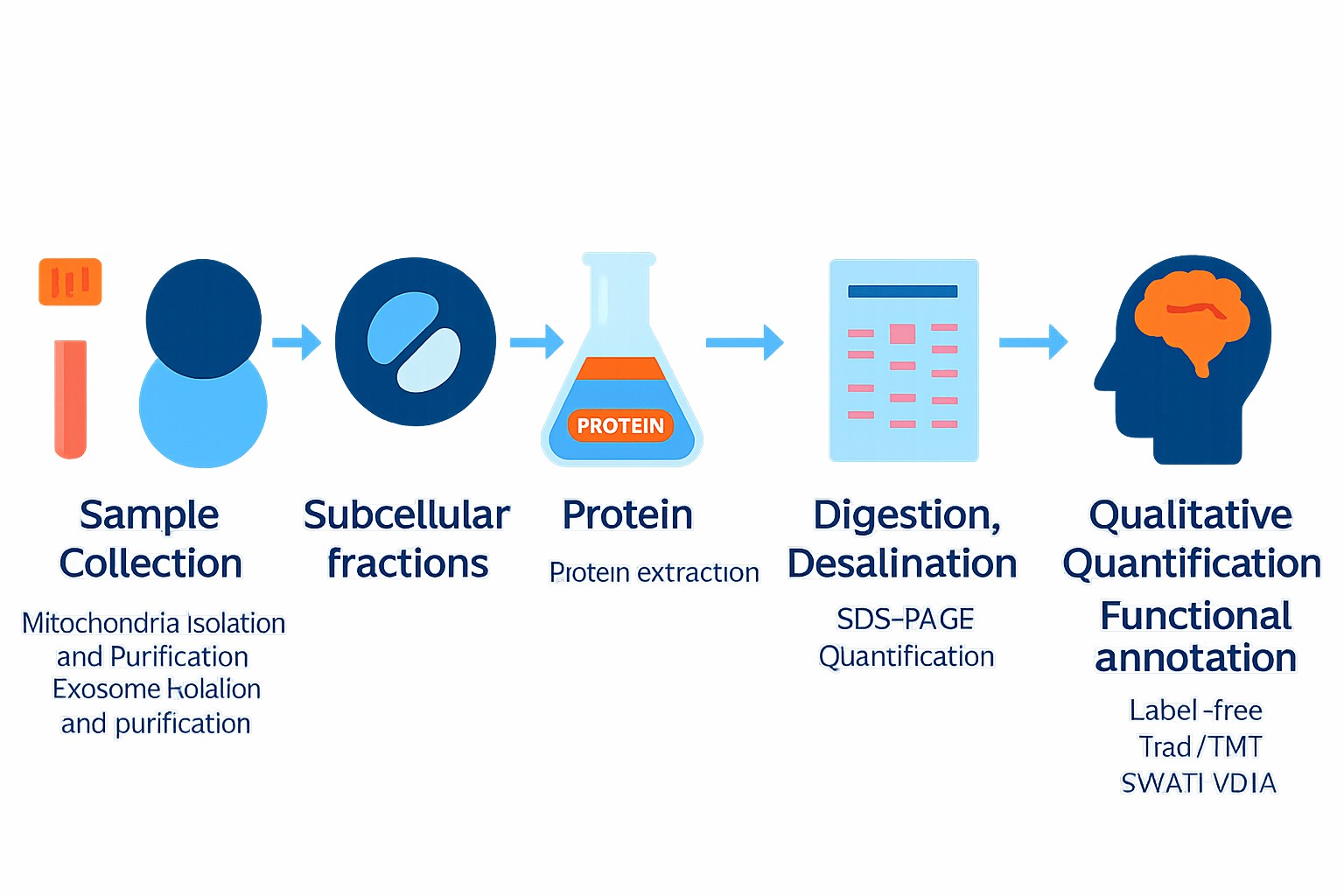
Technical Advantages of Our Proteomic Analysis
We've optimized every step to ensure that our subcellular proteomics service delivers maximum value:
Superior Extraction Efficiency
Our proprietary methods increase protein purity by 10–20% and boost overall protein yield by 15%.
High-Sensitivity Mass Spectrometry
The integration of SWATH data acquisition allows confident identification of proteins at very low abundance levels.
Integrated Database Analysis
Cross-referencing multiple high-quality protein databases improves localization accuracy and expands identification coverage.
Multi-Layered Data Interpretation
We combine experimental data with computational predictions for subcellular localization, enhancing insights into protein functions and cellular pathways.
In addition, Creative Proteomics offers advanced bioinformatics solutions, including:
- Functional annotation and enrichment analyses
- Clustering and network analyses
- Robust statistical evaluations
If you're planning a subcellular proteomics project or wish to explore how our services fit into your workflow, feel free to contact us. We're ready to help you achieve clearer, more actionable proteomic insights.
Technical Platform
| Category | Instrumentation & Software | Why it matters |
|---|---|---|
| High-resolution MS | Thermo Orbitrap Fusion Lumos Tribrid Thermo Q Exactive HF-X Bruker timsTOF Ultra 2 / timsTOF Pro 2 SCIEX TripleTOF 6600 (for SWATH-DIA) | Sub-ppm mass accuracy, >240 k resolution, scan rates up to 40 Hz capture fast nanoLC peaks and low-abundance spectra |
| Nano-LC systems | Thermo UltiMate 3000 and EASY-nLC 1200 | Stable low-flow gradients for single-organelle samples |
| Fractionation / enrichment | Beckman Optima MAX-XP & L‐100K ultracentrifuges; Bio-Comp gradient station; magnetic-bead affinity kits | Yield organelles at ≥90 % purity before MS |
| Data analysis | MaxQuant, Proteome Discoverer, Spectronaut™, Perseus, Cytoscape | Integrated identification, LFQ/SILAC/TMT quant, localization scoring, network & pathway enrichment |
These platforms—operated by PhD proteomics specialists—ensure every project receives uncompromised depth, accuracy, and interpretability.
Sample Requirements
| Sample type | Recommended sample size | |
|---|---|---|
| Animal tissues | Hard tissues (bones, hair) | 300-500mg |
| Soft tissues (leaves, flowers of woody plants, herbaceous plants, algae, ferns) | 200mg | |
| Plant tissues | Hard tissues (roots, bark, branches, seeds, etc.) | 3-5g |
| Microbes | Common bacteria, fungal cells (cell pellets) | 100μL |
| cells | Suspension/adherent cultured cells (cell count/pellet) | >1*107 |
| Fluids | Plasma/serum/cerebrospinal fluid (without depletion of high abundance proteins) | 20μL |
| Plasma/serum/cerebrospinal fluid (with depletion of high abundance proteins) | 100μL | |
| Follicular fluid | 200μL | |
| Lymph, synovial fluid, puncture fluid, ascites | 5mL | |
| Others | Saliva/tears/milk | 3-5mL |
| Culture supernatant (serum-free medium cannot be used) | 20mL | |
| Pure protein (best buffer is 8MUrea) | 300μg | |
| FFPE | Each slice: 10µm thickness, 1.5×2cm area | 15-20 slices |
Customer Case Study: Mapping SPB Interactors in Neurospora crassa

Astrid N. Espino-Vázquez, Rosa Ramírez Cota et al,.Protein interactors of Spindle Pole Body (SPB) components and septal proteins in fungus Neurospora crassa: A mass spectrometry-based dataset 2023. Data in Brief. https://doi.org/10.1016/j.dib.2023.109980
- Project Background
- Client Challenges
- How We Helped
- Results
- Research Impact
- Our Key Advantages
The Spindle Pole Body (SPB) serves as the primary microtubule-organizing center in fungi, essential for accurate cell division and septum formation. A research group from CICESE and KIT aimed to map SPB-associated proteins in Neurospora crassa using advanced subcellular proteomics. Their objective: isolate four tagged SPB components and identify interacting proteins with high confidence.
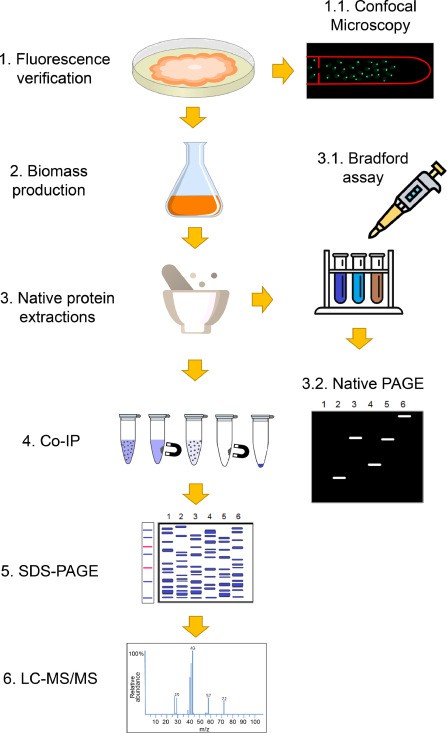 Protein-protein interactions of γ-tubulin (main MTOC component), MZT-1 (integral MTOC microprotein), APS-2 (MTOC and septal protein), and SPA-10 (pore septal protein) were pull-down by Co-IP and identified by LC-MS/MS using the experimental design depicted
Protein-protein interactions of γ-tubulin (main MTOC component), MZT-1 (integral MTOC microprotein), APS-2 (MTOC and septal protein), and SPA-10 (pore septal protein) were pull-down by Co-IP and identified by LC-MS/MS using the experimental design depicted
- Low-abundance proteins: SPB components are typically expressed at very low levels, making detection difficult.
- High background noise: Whole-cell extracts include thousands of unrelated proteins.
- Precision targeting required: SPB and septal proteins must be isolated with compartmental specificity.
Creative Proteomics provided a full-service subcellular proteomics workflow:
- Subcellular Enrichment
- SPB components (γ-tubulin-sGFP, MZT-1-sGFP, APS-2-dRFP, SPA-10-sGFP) were affinity-purified via magnetic GFP/RFP beads.
- Fractionation & LC-MS/MS
- Purified complexes were separated via SDS-PAGE, digested, and analyzed on a Thermo Q Exactive HF nano-LC-MS/MS platform (Mendeley Dataset).
- Bioinformatics & Quantitation
- MS data were searched against the N. crassa proteome, controls subtracted, and quantitative metrics obtained using MaxQuant software.
- Over 1,100 unique proteins were identified across the four SPB components.
- Numerous novel protein interactors were discovered, elucidating connections between SPB and septal pore formation.
- Results were sufficiently robust for publication and functional follow-up.
- Constructed a detailed SPB interactome for N. crassa, offering the first system-level insight.
- Advanced understanding of fungal cell-cycle regulation and septal development.
- Generated high-confidence data ready for downstream validation or publication.
- Expertise in subcellular fractionation and affinity capture
- Access to high-resolution mass spectrometry suitable for low-abundance proteins
- Full bioinformatics support: quantitative analysis, interaction mapping, functional annotation
Frequently Asked Questions
What sample types are compatible with subcellular proteomics?
We work with diverse materials including cell lines, tissues (animal or plant), and cellular organelles such as mitochondria, nuclei, chloroplasts, and exosomes. If you're working with a unique bio-sample, our team will provide tailored guidance.
How much material do I need to submit?
We typically require enough protein to visualize on a gel or proceed with in-solution digestion. For organelle-rich samples like mitochondria or nuclei, 20–50 µg of total protein is usually sufficient—but we work effectively with lower amounts, depending on your samples' quality.
Do you support both gel-based and solution-based proteomics?
Yes.
- Gel-based (GeLC-MS/MS) is ideal for IP or pull-down samples where you want to exclude high-abundance proteins.
- Solution-based digestion is preferred for whole-organelles or membrane preparations, enabling deeper coverage and better quantitation with nano LC MS/MS.
Can you isolate and analyze multiple organelles in one study?
Absolutely. We offer sequential fractionation workflows to separate and analyze organelles like mitochondria, chloroplasts, nuclei, membranes, or exosomes—allowing side by side proteome comparisons across subcellular compartments.
How do you ensure the purity of organelle preparations?
We apply differential and density-based centrifugation, followed by quality checks such as SDS-PAGE, marker protein verification, or Western blotting. These steps help confirm successful compartment purification before mass spectrometry analysis.
What mass spectrometry platforms and quantitation methods are used?
We utilize Thermo's Orbitrap Fusion Lumos or Q Exactive high-resolution instruments. Quantitation methods include Label-free DIA/SWATH, TMT or iTRAQ labeling, and SILAC, depending on your study's needs.
What bioinformatics support do you provide?
Our analysis covers protein identification, quantification, subcellular localization assignment, interaction network mapping, and functional annotation. We also generate visual outputs such as heatmaps, volcano plots, and interaction diagrams to support your interpretation.
Can you highlight compartment-specific proteins or interactions?
Yes. Using quantitative mass spectrometry and controls, we can identify proteins enriched in a specific organelle, map protein complexes, and compare localization patterns across subcellular fractions.
Will I receive raw data and analyzed results?
Absolutely. You'll receive raw MS files, processed identification and quantification tables, and a technical report. The report includes QC metrics, spectral examples, visualization charts, and expert commentary to help interpret findings.
Do you help with data interpretation or publication support?
Yes. We assist with data interpretation, figure preparation (e.g., PCA, heatmaps), and generation of publication-ready materials like annotated protein interaction networks. You also have the option for a consultation to discuss findings and next steps.
Learn about other Q&A about proteomics technology.
Publication

- Regulation of cardiac ferroptosis in diabetic human heart failure: uncovering molecular pathways and key targets. Cell Death Discovery. https://doi.org/10.1038/s41420-024-02044-w
- MS-CETSA functional proteomics uncovers new DNA-repair programs leading to Gemcitabine resistance. Research Square.2024. https://www.researchsquare.com/article/rs-4820265/v1
- Protein interactors of Spindle Pole Body (SPB) components and septal proteins in fungus Neurospora crassa: A mass spectrometry-based dataset. Data in Brief. 2024. https://doi.org/10.1016/j.dib.2023.109980
- Pan-lysyl oxidase inhibition disrupts fibroinflammatory tumor stroma, rendering cholangiocarcinoma susceptible to chemotherapy. Hepatology Communications. 2024. https://doi.org/10.1097/HC9.0000000000000502












