Protein Sequence Analysis Service
Protein sequence analysis is the process of studying the features, function, structure, or evolution of a protein or peptide sequence using various analytical methods. Common techniques include sequence alignment, database search comparisons, and other methods. Through these analyses, the function, molecular structure, and evolutionary relationships of proteins can be revealed. Creative Proteomics can provide several protein sequence analysis services to support your needs.
Submit Your Request Now
×
- Overview
- How It Works
- Our Serives
- Applications
- Advantages
- FAQs
What Is Protein Sequence Analysis?
Protein Sequence Analysis is the process of subjecting a protein or peptide sequence to one of a wide range of analytical methods to study its features, function, structure, or evolution. Methodologies used include sequence alignment, searches against biological databases, and other methods. Since the development of methods of high-throughput production of protein sequences, the rate of addition of new sequences to the databases increased exponentially. Such a collection of sequences does not, by itself, increase the researcher's understanding of the biology of organisms. However, comparing these new sequences to those with known functions is an important way of studying the biology of an organism from which the novel sequence comes. Thus, protein sequence analysis can be used to assign function to proteins by the study of the similarities between the distinct sequences. Nowadays, many tools and techniques are available to analyze the alignment product and provide the sequence comparisons to study its biology.
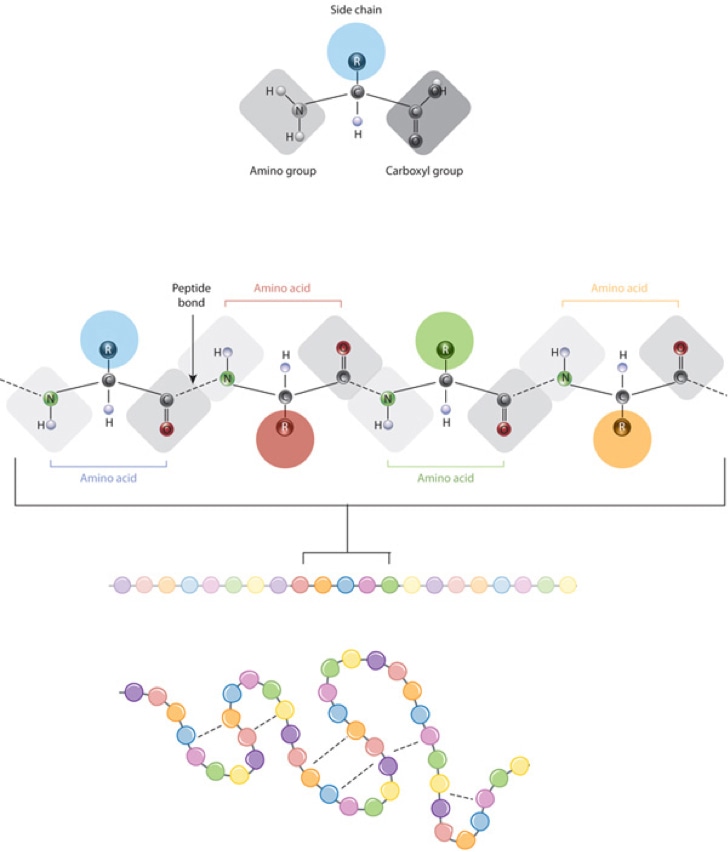 Figure 1. The correlation between amino acid side chain properties and protein structure.
Figure 1. The correlation between amino acid side chain properties and protein structure.
Protein Sequence Analysis in Bioinformatics
Protein sequence database
The completeness and accuracy of protein sequence information in proteomic databases are critical factors influencing the reliability of protein identification and quantification. Ideally, the database should encompass all potential protein sequences present in the sample while excluding unrelated sequences. However, as the exact composition of the sample is unknown prior to analysis, it is common practice to use the complete proteome database of the sample's species for database searches. For samples derived from multiple species, such as those involving viral or bacterial infections, the database should include protein sequences from all relevant species wherever possible.
Table 1 .Commonly used mass spectrometry database
| Name | Description | Website |
|---|---|---|
| UniProt | A widely used protein database integrating Swiss-Prot, TrEMBL, and PIR-PSD. It includes protein sequences obtained post-genome sequencing and biological function information from literature. | https://www.uniprot.org/ |
| Human Protein Atlas (HPA) | Provides information on the tissue and cellular distribution of thousands of human proteins. Includes sections for Cell, Tissue, Pathology, Brain, Blood, and Metabolism, showcasing protein expression in various contexts. | https://www.proteinatlas.org/ |
| String-db | A database for known and predicted protein-protein interactions, widely used in interaction studies. | https://string-db.org/ |
| Human Proteome Map (HPM) | Indicates protein expression and distribution in tissues based on mass spectrometry data. | http://www.humanproteomemap.org/index.php |
| InterPro | A comprehensive database integrating information on protein domains, families, and functional sites from various sources. | http://www.ebi.ac.uk/interpro/ |
Protein sequence analysis software
In proteomics research, database searching involves using computational software to analyze raw mass spectrometry data, enabling the identification and quantification of peptides and proteins.
Commonly used database search software can be categorized into two types. The first type is developed by commercial companies, such as Proteome Discoverer, Spectronaut, Mascot, and ProteinPilot. These tools are highly functional, well-suited to specific mass spectrometers, and easy to operate, though they often come with significant licensing fees.
The second type is created by research groups, including tools like X!Tandem, pFind, Comet, and MaxQuant. These are generally available for free and feature diverse search algorithms, each with unique strengths. However, some may have limitations in specific aspects of performance, and their operation can be more complex. Among these, MaxQuant, developed and continuously refined by the team of Jürgen Cox and Matthias Mann at the Max Planck Institute, has become one of the most widely used tools in proteomics due to its extensive optimization and ongoing updates.
 Figure 2. Commonly used proteomics library search software
Figure 2. Commonly used proteomics library search software
Protein sequence analysis algorithm
- Bayesian Network method
- Support Vector Machine
- Hidden Markov Model
- Artificial Neural Network method
- Regression Analysis
- Alignment-free sequence analysis
- Other methods if you need!
Our Service
The protein mass spectrometry data analysis services provided by Creative Proteomics include database searching, data quality control, data preprocessing, statistical analysis, functional enrichment analysis, and personalized analysis, among others. The protein sequence analysis module offers the following services:
- Amino acid composition analysis
- Signal peptide prediction
- Protein motif prediction
- Protein ligand binding site prediction
- Transmembrane prediction
The associated protein sequencing services:
Protein Identification Services
- iTRAQ-based Proteomics Aanalysis
- SILAC-based Proteomics Analysis Service
- Label-free Quantification Service
- TMT Based Proteomics Service
- Absolute Quantification (AQUA)
- Semi-quantitative Proteomics Analysis
- Parallel Reaction Monitoring (PRM)
- SRM & MRM
Applications of protein sequence analysis
Sequence Similarity Analysis
The comparison of sequences in order to find similarity, often to deduce if they are homologous.
Variant Detection
Identification of sequence differences and variations.
Structure Prediction
Identification of molecular structure from sequence alone.
Feature Annotation
Identification of intrinsic features of the sequence, such as active sites, PTM sites, gene-structures, and regulatory elements.
Evolutionary Analysis
Revealing the evolution and protein diversity of sequences and organisms.
Our Advantages
- Comprehensive Database Integration: We leverage top protein databases like UniProt and Human Protein Atlas for precise sequence alignment and functional annotation.
- Advanced Data Analysis: Using industry-standard software (e.g., MaxQuant, Proteome Discoverer), we ensure efficient protein identification and quantification.
- Predictive Analysis: We provide insights through signal peptide, motif, and ligand binding site predictions.
- Tailored Solutions: Our customized analysis meets specific research needs with precise results.
- Wide-ranging Applications: We offer solutions for sequence similarity, variant detection, structure prediction, and evolutionary analysis.
Protein Sequence Analysis FAQs
What are the key methods used for protein sequence alignment?
Protein sequence alignment is typically performed using two primary methods: pairwise and multiple sequence alignment. Pairwise alignment compares two sequences to find the best match, using algorithms like Needleman-Wunsch or Smith-Waterman. For multiple sequence alignment, tools like ClustalW and MUSCLE are used to align more than two sequences, helping to identify conserved regions and evolutionary relationships. These methods can reveal homology, conserved motifs, and structural domains that are essential for understanding protein function and evolutionary history.
How do you handle incomplete or ambiguous protein sequence data?
Incomplete or ambiguous protein sequence data can be challenging in sequence analysis, but several strategies can help address these issues. In cases where parts of the sequence are missing, homology-based approaches are often used to fill in gaps, aligning the incomplete sequence to closely related proteins in well-curated databases like UniProt. In cases of ambiguous sequence data, computational methods such as machine learning algorithms and probabilistic models (e.g., Hidden Markov Models) can be applied to predict the most likely sequence or structural conformation. Additionally, experimental techniques like mass spectrometry and NMR spectroscopy can be used to verify or supplement sequence data, improving the reliability of the analysis.
What is the importance of variant detection in protein sequence analysis?
Variant detection is crucial in protein sequence analysis, especially for understanding genetic mutations that lead to disease. By comparing the protein sequences from healthy and diseased individuals, researchers can identify mutations, substitutions, deletions, or insertions that may cause dysfunction or contribute to disease phenotypes. This process is often used in personalized medicine to tailor treatments based on individual genetic profiles. Variant detection also helps in identifying drug resistance mutations, understanding disease progression, and discovering new therapeutic targets, making it a vital aspect of protein analysis in clinical and translational research.
Learn about other Q&A about other technologies.







