Metabolite Identification and Confidence Levels
Defined by the Metabolomics Standards Initiative, there are four levels of metabolite identification confidence: confidently identified compounds (level 1), putatively annotated compounds (level 2), putatively annotated compound classes (level 3) and unknown compounds (level 4). Definitive (level 1) identification requires comparing the two or more orthogonal properties such as retention time, m/z, and fragmentation mass spectrum for the metabolite of interest to the same properties of an authentic chemical standard observed under identical analytical conditions. Putative (level 2 or 3) annotation basically relies on only one or two properties and often based on comparison to data collected in different laboratories and acquired with different analytical methods, rather than a direct comparison with an authentic chemical standard under the same analytical conditions.
Characteristics of Unknown Metabolites
An unknown metabolite is a small molecule that can repeatedly be detected but whose chemical identity has not been identified yet. Though unidentified and unclassified, these compounds can still be differentiated and quantified in a metabolomics experiment based upon spectral data. In LC-MS experiment, an unknown compound would be defined by a unique retention time, one or multiple masses, or a specific fragmentation pattern of the primary ions. In NMR experiment, an unknown compound would be defined by a pattern in the chemical shifts. Unknowns may represent previously undocumented small molecules, such as secondary products of metabolism or rare xenobiotics, or they may constitute molecules from established pathways but could not be assigned with current libraries of NMR reference spectra or MS fragmentation patterns.
Challenges in Identifying Unknown Compounds
Identification of unknown compounds is labor-consuming and cost-intensive, often requiring preparative scale isolation for NMR studies or extensive chemical synthesis enabling structural comparisons using MS/MS. Therefore, unsurprisingly, most reports focus on the detection of metabolites with available authentic commercial standards or at least existing in metabolite databases. Because of the inherent instability of many metabolites and lack of demand, only a few thousand commercially analytical standards are available. However, these standards and the metabolites existed in databases account for only a portion of endogenous metabolites. Therefore, effective methods for prioritizing, studying, and ultimately identifying uncharacterized metabolites are of great importance for metabolomics study. Successful identification of unknown metabolites will exert a great impact on biomarker discovery and omics-research.
Creative Proteomics' Approach to Identifying Unknown Metabolites
To identify the unknown compounds in metabolomic samples, different groups of metabolites should be discriminated. Metabolites of different nominal mass, metabolites with the same nominal mass but of different molecular formula and monoisotopic mass, and metabolites of the same nominal and monoisotopic masses but with different chemical structures should be discriminated. For example, leucine and isoleucine are isomers with different structures, however with the identical nominal and monoisotopic masses. What's more, since single metabolites can be detected in a mass spectrometer as various derived species, it is important to assign the various derived species to the parent metabolite correctly. For example, in amino acid analysis, the reaction of chemical derivatization by trimethylsilylation (TMS) and amino acids will result in the generation of amino acids containing 1, 2, or 3 TMS groups, which all should belong to the same parent amino acids.
By combining NMR, LC-MS/MS, and rule- and algorithm-based methodologies, Creative Proteomics develops a new strategy to identify unknown metabolites allowing for accurate identification of metabolites in complex biological samples. Creative Proteomics is still on its way to develop the tools and databases and provide integration of these different tools and databases for true and complete identification of all metabolites in an automated manner in the future.
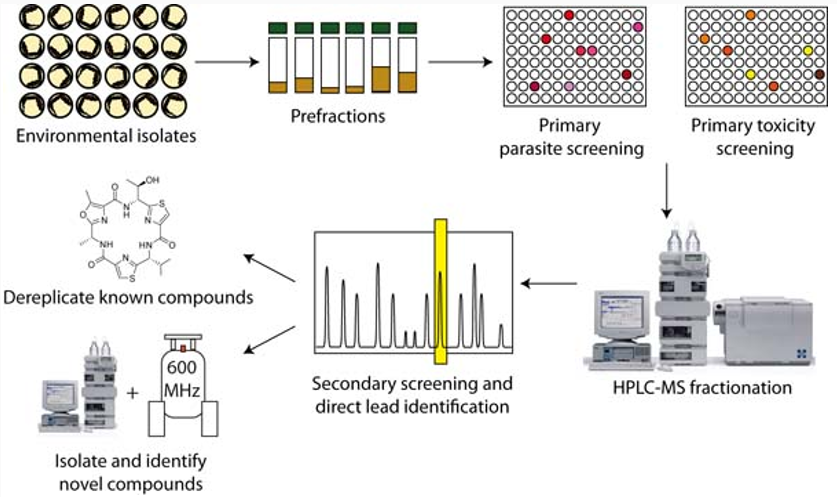
Workflow

Service Advantages of Creative Proteomics in Identifying Unknown Metabolites
Multi-Technology Integration: Creative Proteomics employs a holistic approach to metabolite identification, harnessing a diverse array of techniques including NMR, LC-MS/MS, and rule- and algorithm-driven methodologies. This integrative strategy is meticulously designed to ensure an elevated level of precision and credibility when confronting the enigma of unknown metabolites within intricate biological matrices. The amalgamation of multiple technologies affords a comprehensive vantage point, enabling the exhaustive characterization of metabolites residing in complex biological specimens.
Highly Automated Processes: Creative Proteomics is at the forefront of advancing automation in the realm of metabolite identification. The continuous development of sophisticated tools and extensive databases culminates in the realization of authentic and comprehensive automation, encompassing the identification of all metabolites. This elevated degree of automation expedites the identification process, curtails human intervention, and bequeaths results of remarkable quality.
Tailored Solutions: Acknowledging the inherent distinctiveness of each research venture, Creative Proteomics prides itself on delivering bespoke solutions meticulously crafted to harmonize with the specific requisites of individual clients. Whether the endeavor entails focused investigations of particular metabolite categories or the comprehensive scrutiny of metabolomics across a spectrum of project objectives, Creative Proteomics is adept at curating tailored strategies.
Data Integration and Profound Analysis: The company extends its services to encompass data integration and intricate analyses, empowering clients to gain an enhanced grasp of intricate metabolomics datasets. This analytic prowess not only facilitates the identification of potential biomarkers but also deepens the comprehension of biological specimens, thereby fostering the progression of metabolomics research.
Seasoned Scientific Expertise: At the heart of Creative Proteomics lies a cadre of seasoned scientists endowed with specialized acumen and competencies within the metabolomics domain. These experts not only furnish impeccable experimental data but also proffer sagacious guidance pertaining to the identification and interpretation of metabolites.
Punctual Project Delivery: Creative Proteomics steadfastly pledges to timely project delivery, aligning with the exacting schedules of clients. The organization's streamlined workflow optimization efficaciously mitigates project turnaround times, exemplifying their unwavering commitment to punctuality.



















