Small-Molecule/Protein Complex Native MS Service
Direct, label-free detection of small-molecule binding to protein targets via native mass spectrometry.
Understanding how small molecules interact with their protein targets is the foundation of drug discovery. Whether you are validating hits from a high-throughput screen, confirming fragment binding in an FBDD campaign, or characterizing the mechanism of action of a lead compound, you need direct evidence of binding — not an inference from an indirect assay.
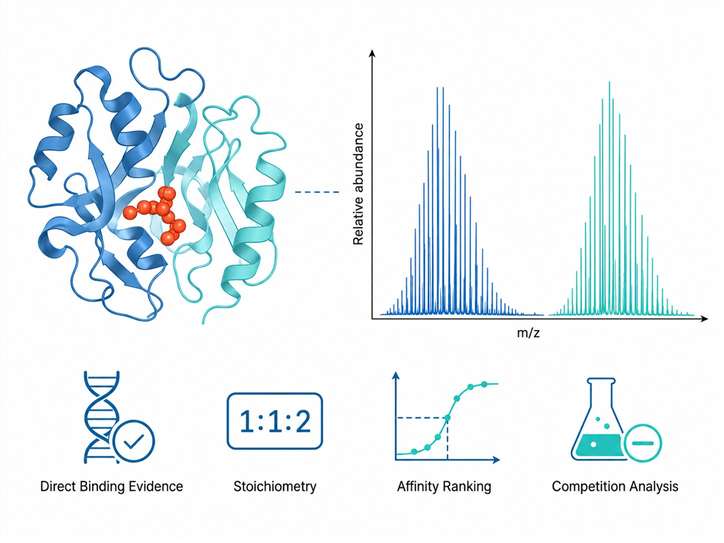
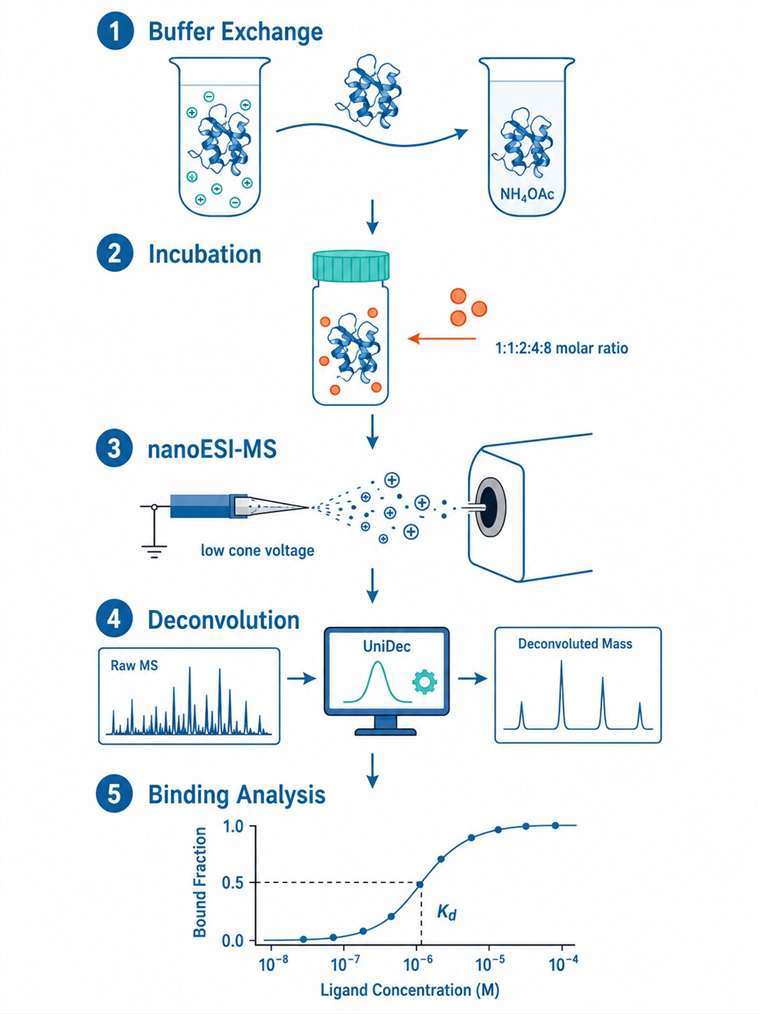
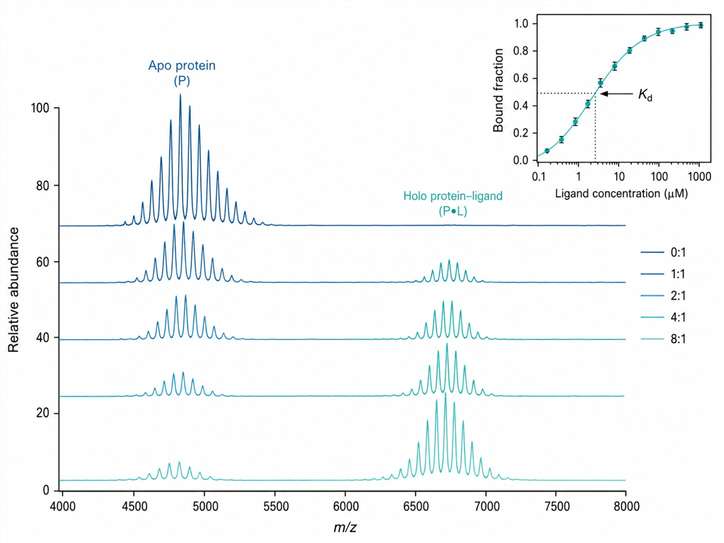
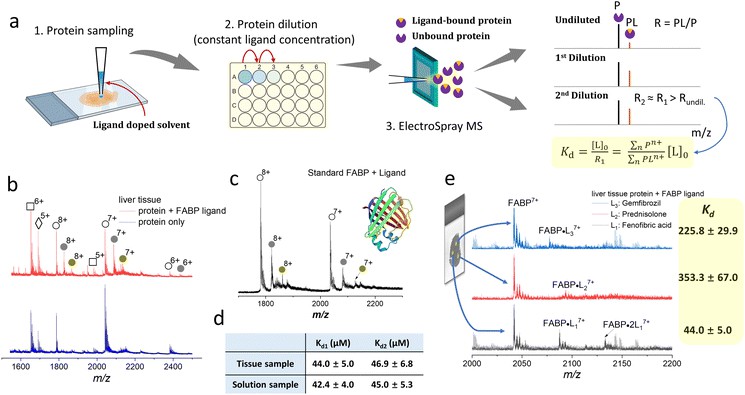
Our Small-Molecule/Protein Complex Native MS Service provides that direct evidence. Using electrospray ionization under carefully controlled native conditions, we preserve non-covalent protein-ligand complexes intact through the mass spectrometry analysis. The result is unambiguous mass-based confirmation of complex formation, binding stoichiometry, and relative binding affinity — all from picomoles of protein, in minutes per sample.
Why researchers choose this service:
- Direct mass evidence of complex formation — no labels, no immobilization, no inference
- Binding stoichiometry from a single experiment — 1:1, 2:1, or higher-order complexes
- Relative affinity ranking and Kd estimation from titration series
- Competition binding analysis for multi-ligand studies