Proteomics (DIA/SWATH) Drug-Response Profiling
Unbiased, deep proteome-wide quantification of drug-induced protein expression changes using DIA/SWATH mass spectrometry.
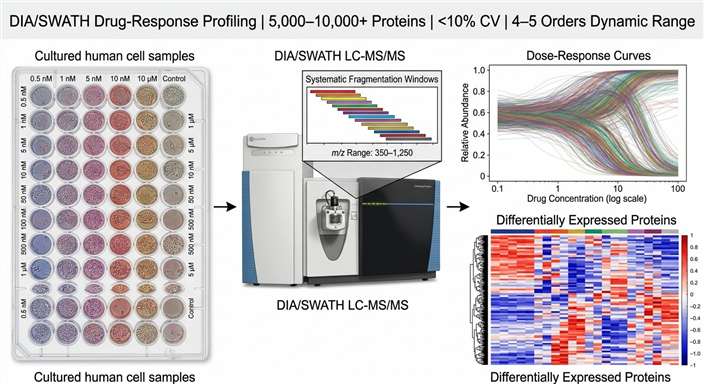
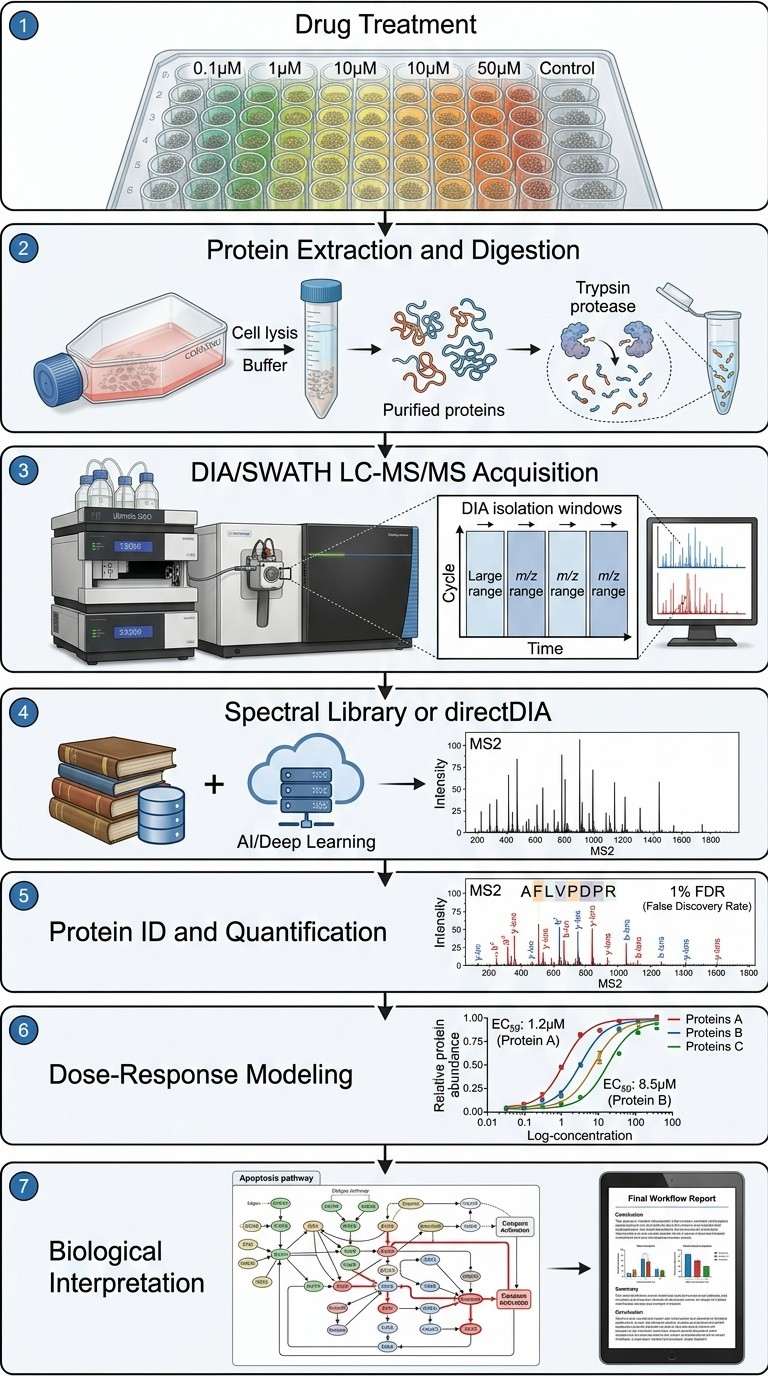
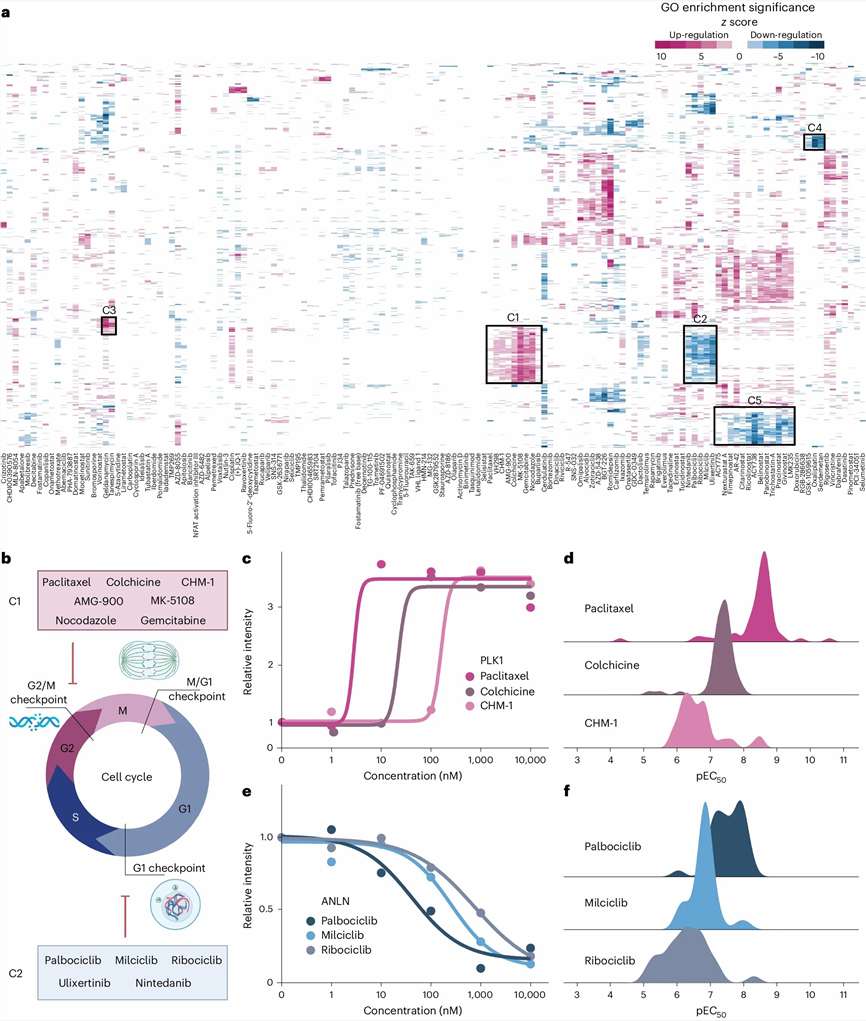
Our DIA/SWATH drug-response profiling workflow captures 5,000–10,000+ proteins per run with inter-run CVs below 15%, giving you dose-response curves for thousands of proteins from a single experiment. The data becomes a permanent digital record you can re-mine for new targets or biomarkers years later.
Key Advantages:
- Deep, unbiased proteome coverage — DIA fragments all detectable peptides systematically, bypassing the stochastic sampling bias of DDA
- Quantitative reproducibility across conditions — consistent CVs below 15% across triplicate runs
- Permanent digital proteome map — re-analyze data for new targets or biomarkers years later
- End-to-end bioinformatics — from raw DIA data to publication-ready figures
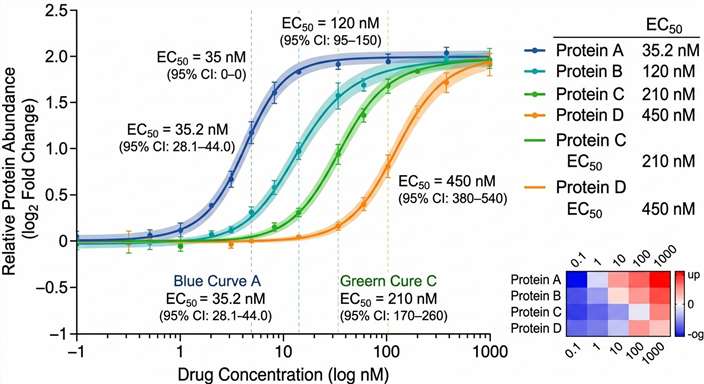
- Dose-response curve fitting with EC50 estimation for thousands of proteins