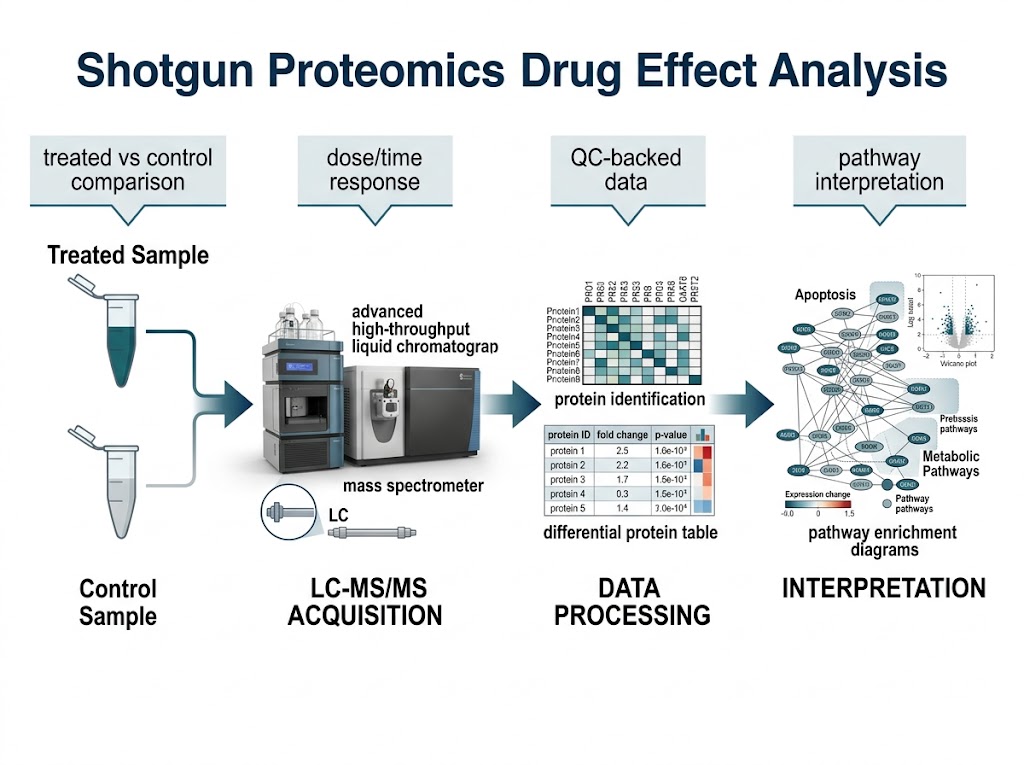
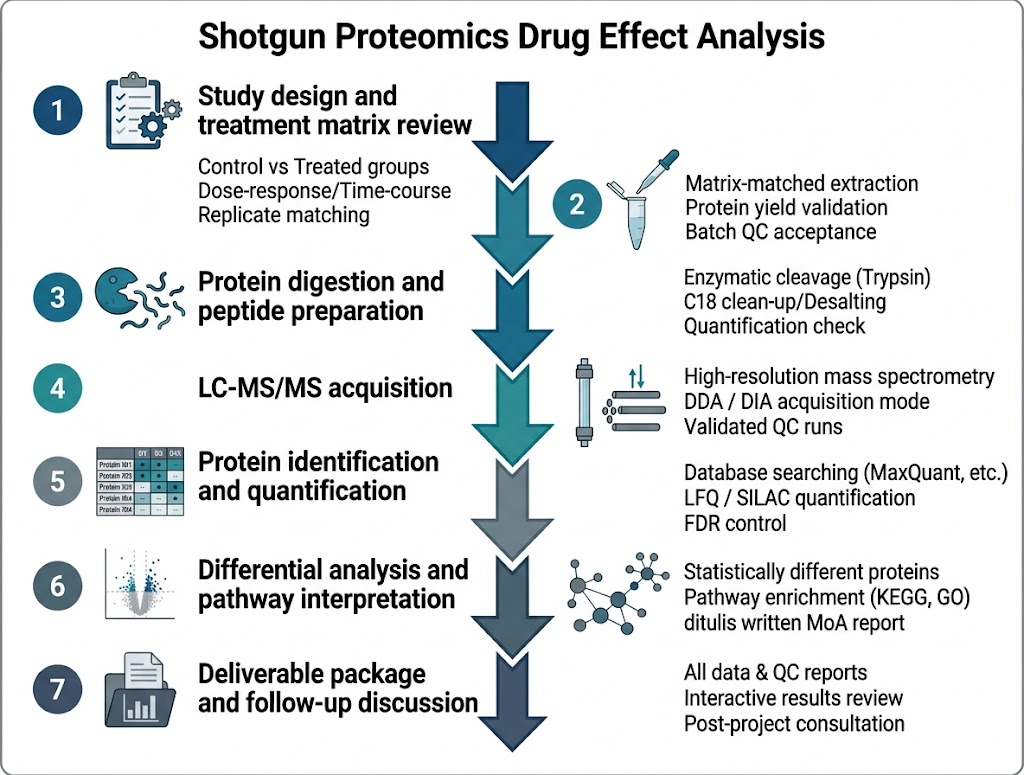
Background
Treatment studies often produce complex biological responses that are difficult to interpret from phenotype alone. In a 2025 study, García-Hernández and colleagues used quantitative proteomics to study molecular mechanisms in a non-Hodgkin lymphoma mouse model treated with incomptine A.
The paper compared treatment-associated proteome changes in lymph node samples and used pathway analysis to interpret altered protein groups. This type of study is relevant to drug-effect proteomics because it shows how protein abundance data can be connected to pathway-level interpretation.
Source: Quantitative Proteomics and Molecular Mechanisms of Non-Hodgkin Lymphoma Mice Treated with Incomptine A, Part II
Methods
The study used treatment groups including vehicle control, methotrexate, and incomptine A at 5 mg/kg and 10 mg/kg. Lymph node pools were prepared according to group and anatomical location.
For proteomics, the researchers extracted proteins, measured protein concentration, reduced and alkylated samples, digested proteins with trypsin, labeled peptides with TMT reagents, combined samples, fractionated peptides, and analyzed them by nano LC-MS/MS. The downstream analysis used KEGG, Reactome, and Gene Ontology databases to interpret protein-level changes.
Results
The study identified and quantified 2717 proteins. Differential expression was evaluated using fold-change thresholds, and the paper reported 412 differentially expressed proteins across treatment comparisons.
- The study compared down-regulated and up-regulated proteins across multiple treatment groups, including incomptine A dose groups and methotrexate.
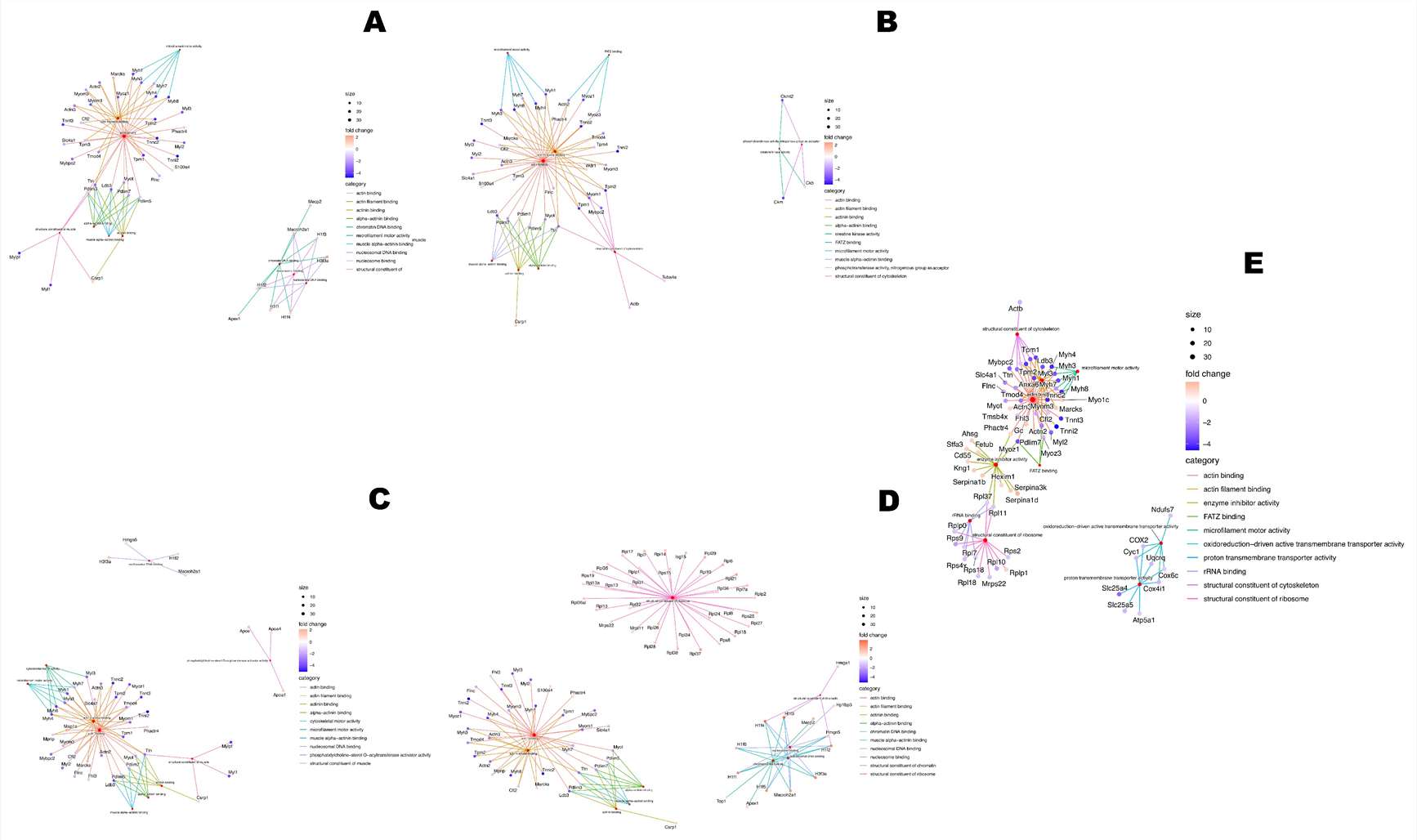
- Figure 4 showed Gene Ontology Molecular Function network enrichment from up-regulated and down-regulated proteins across treatment comparisons.
- In the C− versus 5LANM comparison, the authors reported 76 down-regulated and 69 up-regulated proteins, with altered proteins related to cytoskeleton organization, actin binding, microfilament activity, nucleosome binding, and chromatin DNA binding.
- In the C− versus 5RINM comparison, the authors reported 117 down-regulated and 72 up-regulated proteins, including functional categories related to cytoskeleton structure and phosphotransferase activity.
- In the C− versus 10RINM comparison, the authors reported 83 down-regulated and 132 up-regulated proteins, with altered proteins related to actin binding and ribosome structural components.
- In the C− versus MTX comparison, the authors reported 111 down-regulated and 63 up-regulated proteins, with altered proteins related to proton transmembrane transporter activity and enzyme inhibitor activity.
Figure 4 is useful for this page because it shows how quantitative proteomics can move from protein abundance changes to functional network interpretation. It does not simply list proteins; it organizes altered proteins into molecular function patterns across treatment comparisons.
Conclusion
This case supports shotgun proteomics drug effect analysis because it demonstrates a full path from treatment comparison to protein-level response and pathway interpretation. For drug discovery teams, this type of workflow can help identify treatment-associated functional changes, compare response patterns, and select proteins or pathways for follow-up validation.