Disease Mechanism and Pathway Analysis Service
Multi-modal disease mechanism analysis integrating target engagement, signaling, metabolomics, chemical proteomics, and ADME characterization from the MassTarget platform.
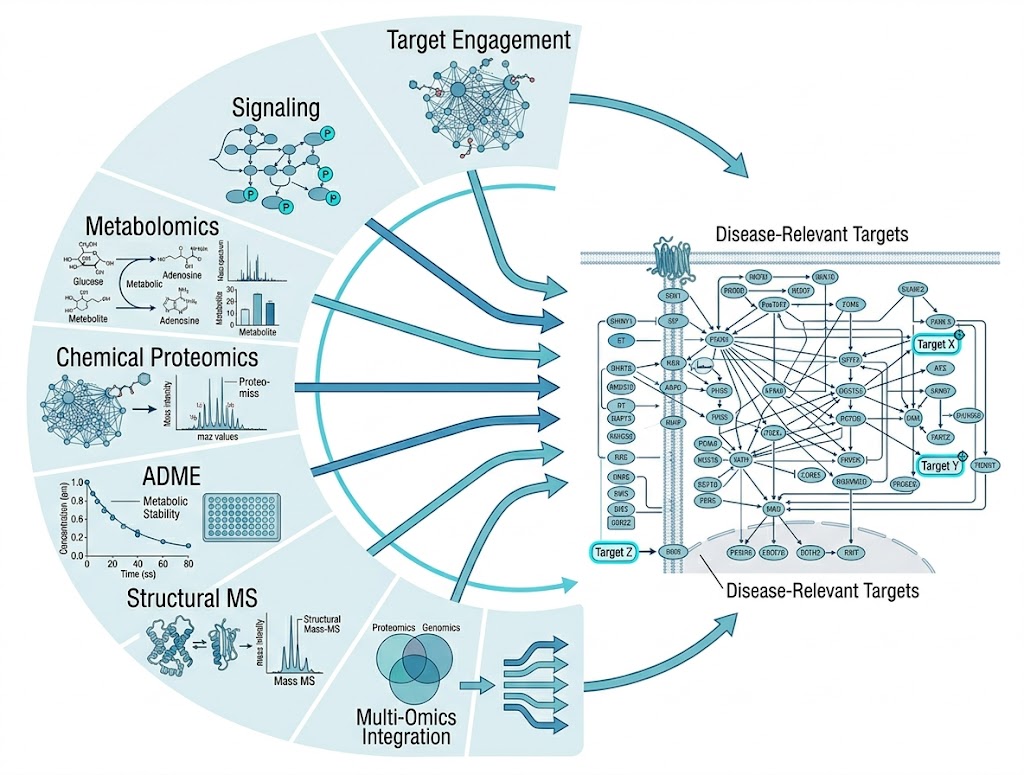
Disease mechanisms operate across multiple biological dimensions — target binding, signaling modulation, metabolic reprogramming, and cellular phenotype. Understanding how a compound or genetic perturbation drives a disease phenotype requires evidence from each of these layers.
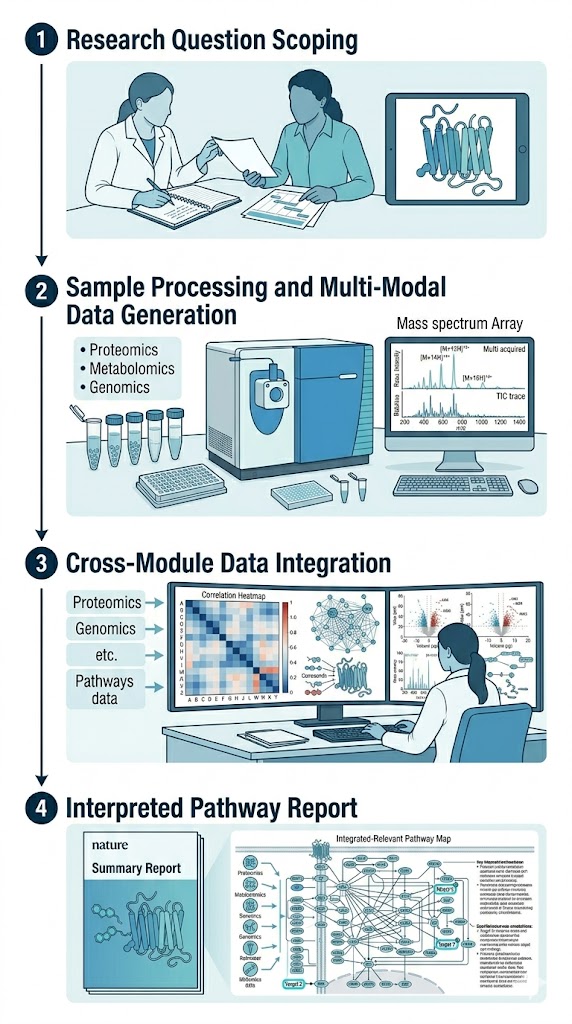
The MassTarget platform addresses this challenge by integrating seven complementary MS-based service categories into a coordinated disease mechanism analysis workflow. From affinity screening and thermal profiling to phosphoproteomics, metabolomics, chemical proteomics, and ADME characterization — each module provides a distinct piece of the mechanism puzzle.
Solution Highlights:
- Seven integrated service categories covering the full mechanism analysis spectrum
- Modular design — select the combination that matches your research question
- Coordinated sample processing for consistent, comparable datasets
- Cross-module integration with interpreted biological pathway reports
- Direct pipeline connectivity to target discovery and validation services