Covalent Labelling Mass Spectrometry Service for Protein Conformational and Binding Analysis
Residue-aware structural comparison for protein accessibility, binding, and conformational change studies.
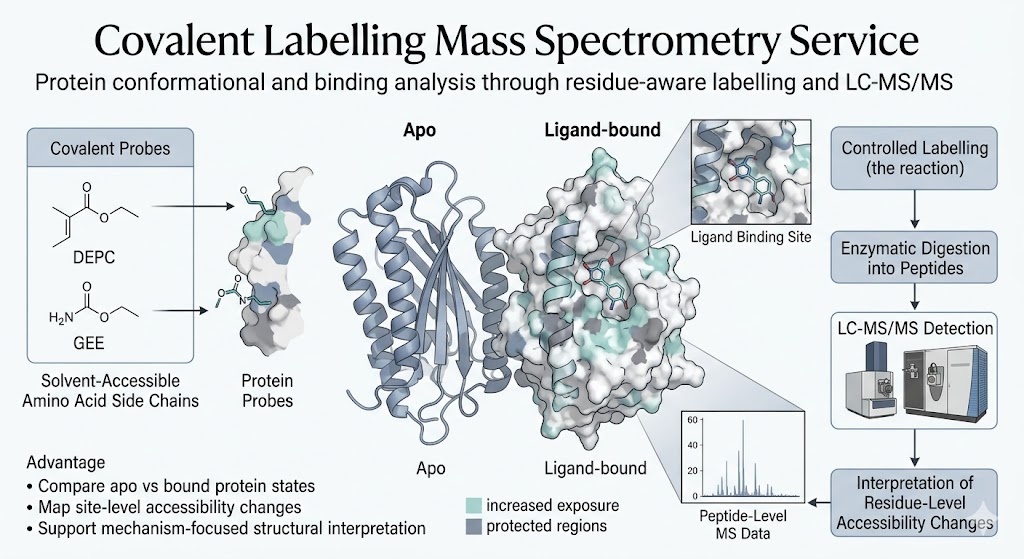
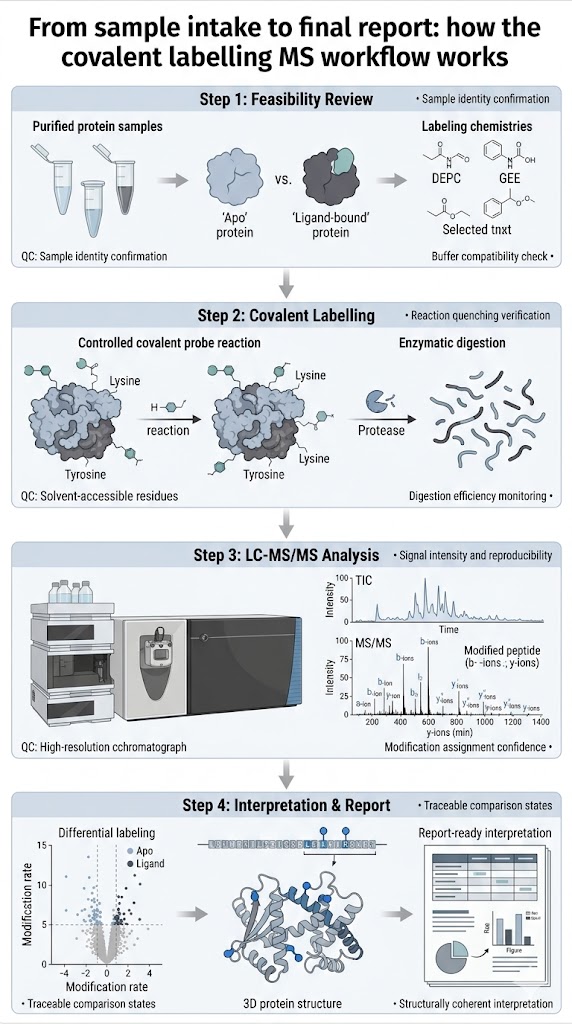
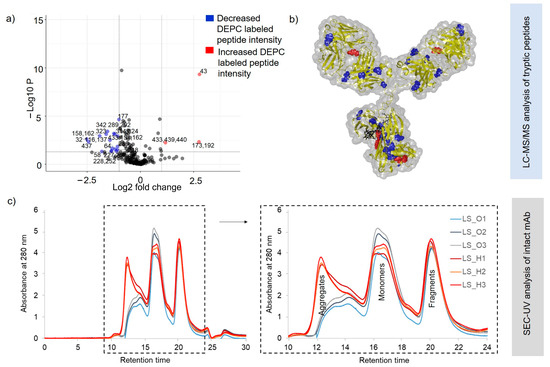
Our Covalent Labelling Mass Spectrometry Service helps you compare protein states through residue-aware labeling and LC-MS/MS analysis. We use covalent footprinting strategies such as DEPC, GEE, and related probes to support protein conformational analysis, binding studies, and site-linked interpretation of structural change.
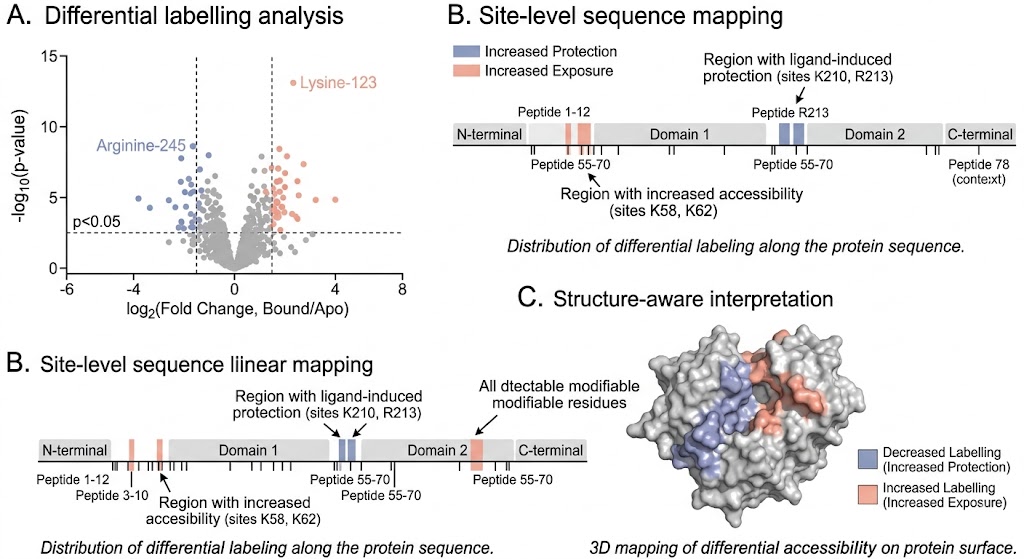
When you need to understand how a protein changes across states, a simple identification result is often not enough. You may need to know which regions become more exposed, which sites become more protected, or whether ligand binding changes solvent accessibility in a way that supports a mechanism hypothesis.
This service is built for that kind of question. We focus on project fit, labeling strategy selection, sample readiness, and reportable outputs that can support scientific review and next-step decision-making.
What we help you evaluate:
- Protein conformational change across defined states.
- Ligand-induced accessibility shifts.
- Apo vs bound structural differences.
- Stress- or formulation-related perturbation.
- Site-linked evidence for mechanism-focused studies.