Epitope Mapping of Human Polyclonal Antibodies to the fHbp Antigen of a Neisseria Meningitidis Vaccine by Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS). https://www.sciencedirect.com/science/article/pii/S1535947624000240
Background
Mapping epitopes for polyclonal antibodies (pAbs)—such as those generated by vaccines or found in convalescent plasma—is notoriously difficult for the biopharmaceutical industry. Because the human immune response produces a highly heterogeneous mixture of antibodies that bind to many different regions of an antigen simultaneously, traditional monoclonal mapping methods (like obtaining a single co-crystal) are completely ineffective. Understanding exactly which parts of a vaccine antigen are targeted by the immune system is vital for evaluating vaccine efficacy and ensuring batch-to-batch structural consistency.
Methods
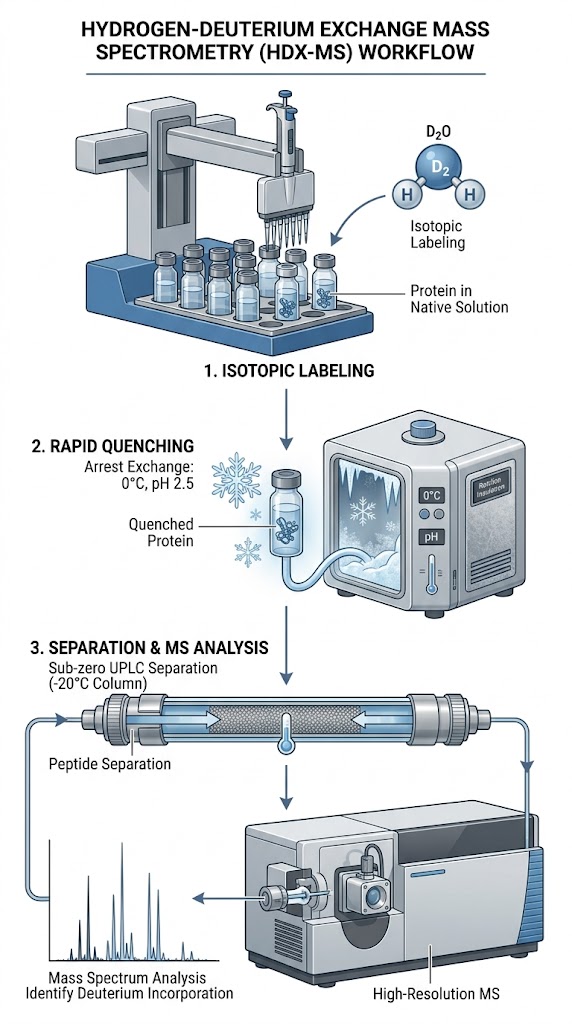
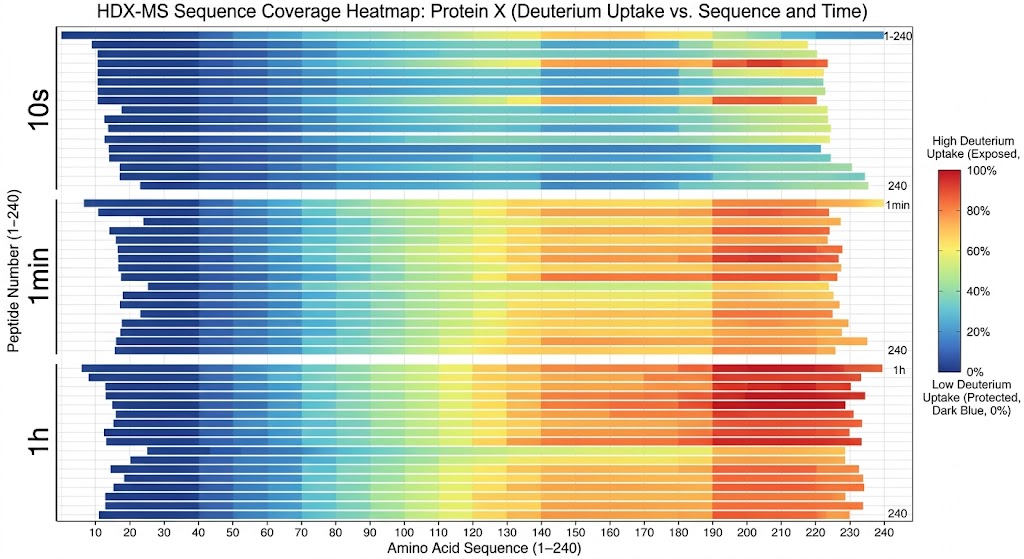
To overcome this massive analytical hurdle, researchers utilized HDX-MS to map the binding interfaces of human pAbs against the fHbp (factor H binding protein) antigen, a critical component of a Neisseria meningitidis vaccine. The recombinant fHbp antigen was incubated with various purified pAb fractions derived from vaccinated individuals. The antigen-antibody complexes were then subjected to timed deuterium exchange (ranging from 10 seconds to 10,000 seconds), rapidly quenched to pH 2.5 to halt the exchange, digested online with pepsin, and analyzed via high-resolution MS to track the localized mass shifts across the entire protein sequence.
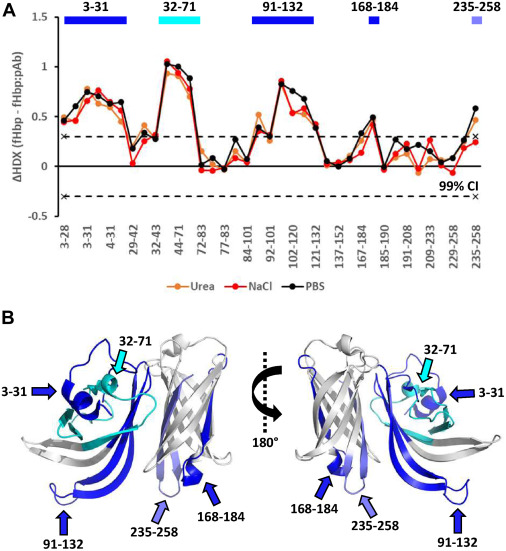
Results
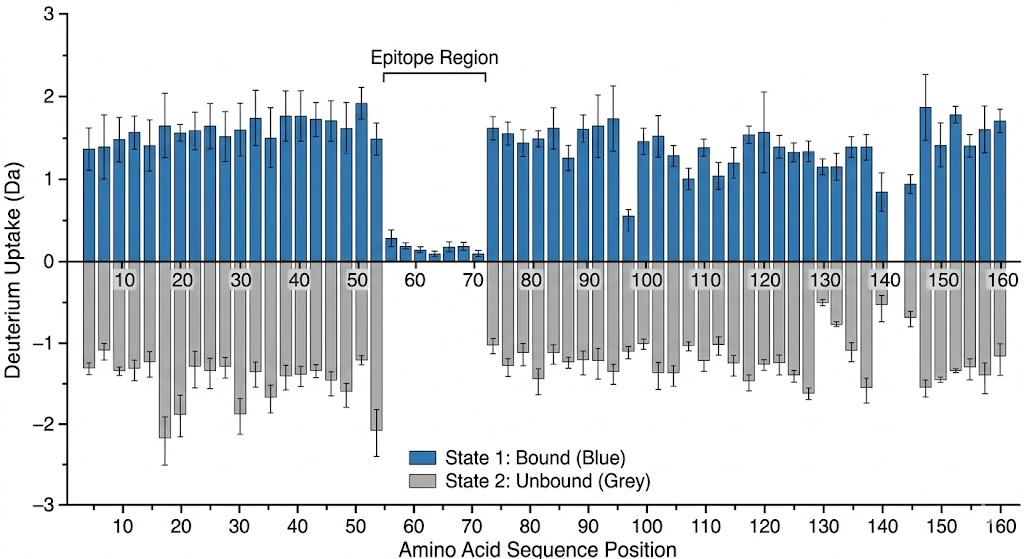
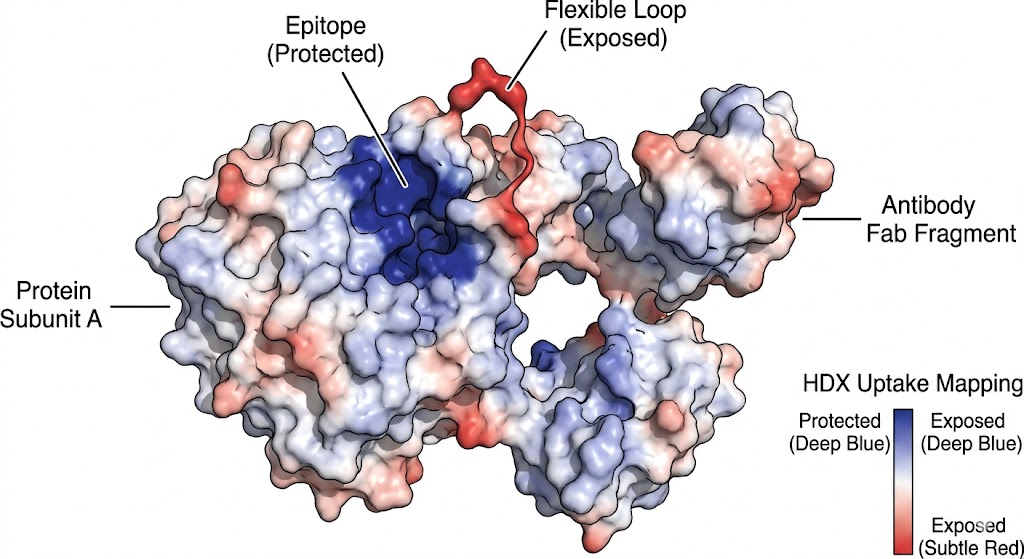
As shown in the graphical data representations from the published study, HDX-MS successfully managed the complexity of the polyclonal mixture. It identified highly specific, localized decreases in deuterium uptake on the fHbp antigen upon pAb binding. The high-resolution mass spectrometry data, coupled with rigorous statistical difference testing, clearly distinguished the immunodominant conformational epitopes across different vaccine recipient samples. Furthermore, it revealed that the polyclonal response was directed at several distinct, spatially separated patches on the folded protein surface.
Conclusion
HDX-MS proved to be a highly effective, label-free method for profiling the complex polyclonal epitope landscape. It provided critical, high-resolution structural insights for vaccine evaluation and development that would be impossible to capture using standard mutagenesis, ELISA, or crystallography. This powerful application highlights HDX-MS as an essential tool not just for monoclonal antibody discovery, but for advanced vaccine characterization and immune response profiling.