Background
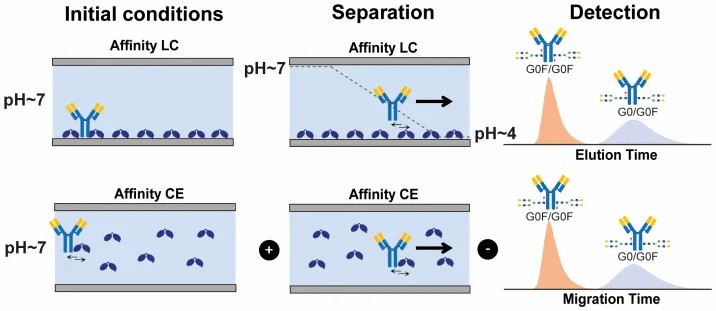
In early-stage biologics and drug discovery, understanding how distinct isoforms or closely related biological species interact with a target is notoriously difficult. Bulk biophysical methods often average out the signals, masking the subtle but critical binding differences between these species. For example, the precise glycosylation state of an antibody can drastically alter its binding affinity to effector receptors, dictating its clinical efficacy and influencing Antibody-Dependent Cellular Cytotoxicity (ADCC).
Methods
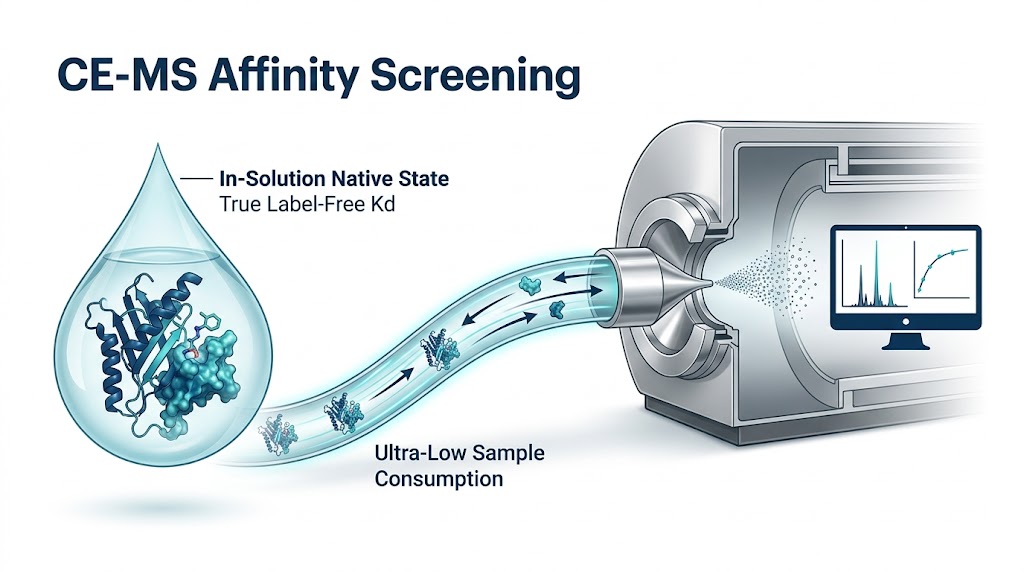
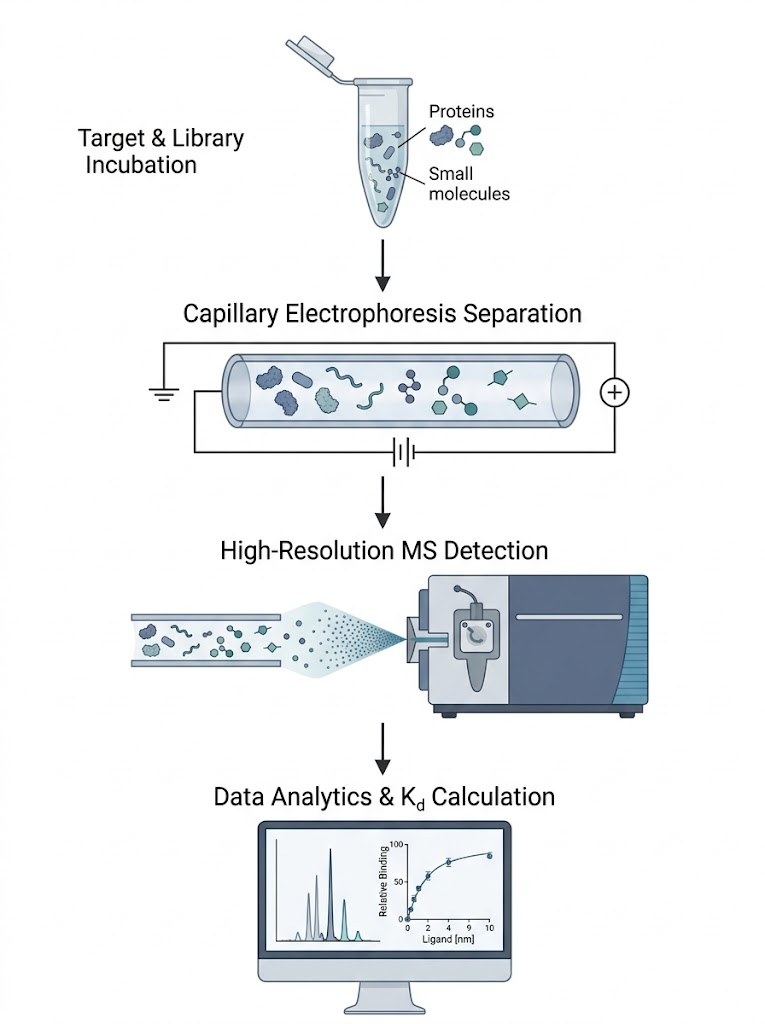
To resolve this, an affinity separation-mass spectrometry workflow was deployed to study the interaction between varying antibody glycoforms and the FcγRIIIa receptor. The target receptor and the heterogeneous antibody ligands were incubated naturally in-solution, allowing them to reach true thermodynamic equilibrium without the steric hindrance of surface tethering. The intact complexes were then separated using high-efficiency capillary techniques. Crucially, they were subsequently analyzed using a high-resolution mass spectrometer to directly resolve the interactions based on exact mass.
Results
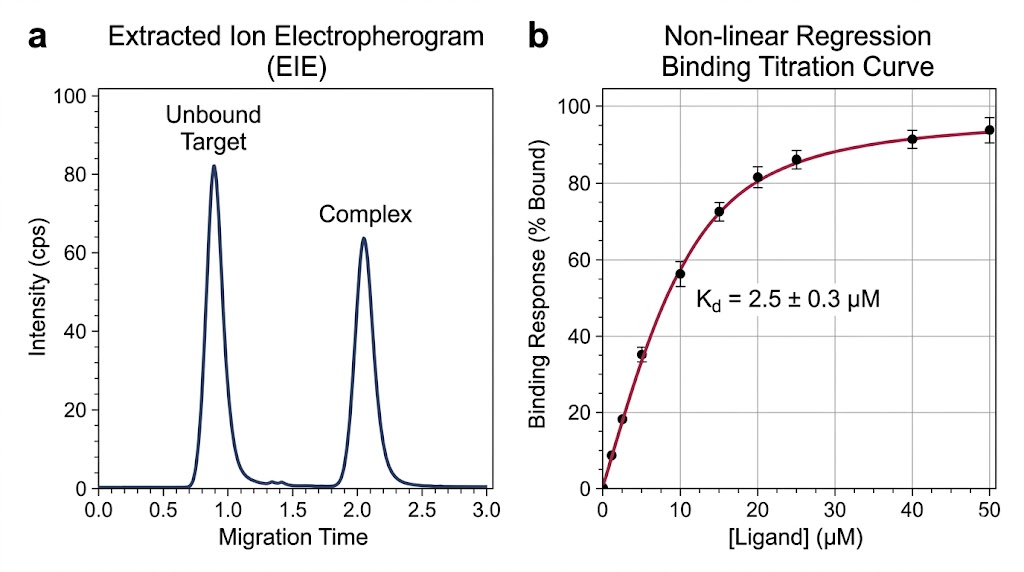
The high-resolution MS readout provided clear, reviewable signal evidence of the distinct interaction profiles without requiring any target modification or fraction purification. Because the mass spectrometer can easily detect the exact mass difference of specific sugar residues (for example, the ~146 Da mass shift indicating the presence of a fucose residue, or the precise mass of terminal galactose units), it could distinguish which specific glycoforms were actively binding to the receptor in real-time. As demonstrated in Figure 1 of the published methodology, the extracted ion chromatograms (EICs) successfully differentiated the specific binding signatures of these closely related species. The data clearly showed that fully fucosylated species had different binding profiles compared to hemi-fucosylated species, and even differentiated binding based on the specific F158/V158 receptor variants, showcasing the platform's unparalleled resolution.
Conclusion
This approach proves that high-resolution affinity separation-MS is highly effective not just for simple binary screening, but for dissecting complex molecular interactions in their native state. By directly observing the exact mass of the binding partners, it provides discovery teams with robust, artifact-free data to prioritize candidates based on highly specific structural nuances that govern in vivo efficacy.