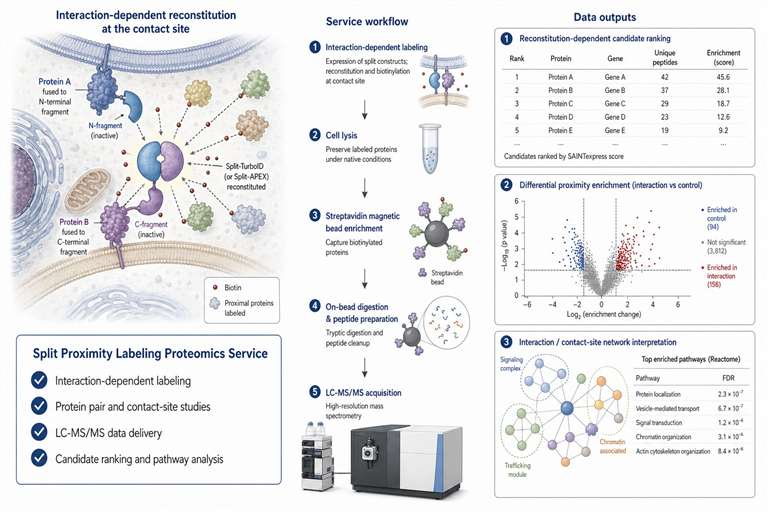
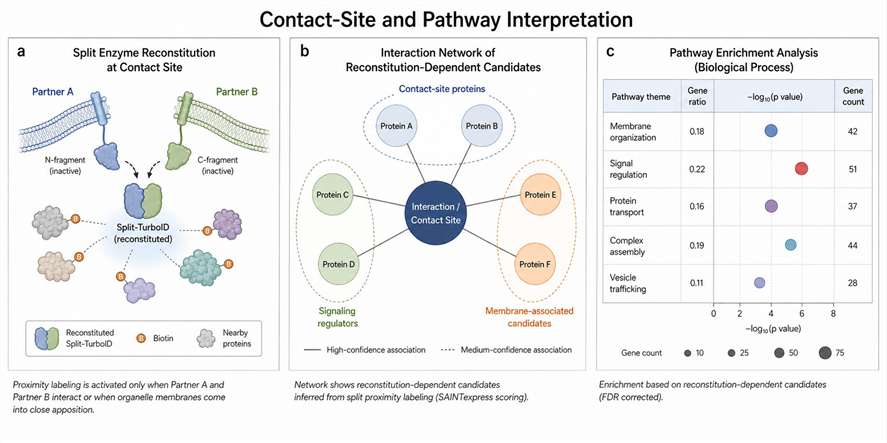
Capture Protein Neighborhoods Only When Specific Interactions Occur
Split proximity labeling uses two inactive enzyme fragments that regain activity only when they are brought close enough by a biological event. In a typical design, one fragment is fused to one protein, organelle marker, or membrane-localized component, while the other fragment is fused to a second partner. When the two partners come together, the split enzyme can reconstitute activity and label nearby proteins.
This design is helpful when the research question is not simply bait-centered, but interaction-centered. You may want to capture proteins near a protein pair only when the pair interacts, proteins near an organelle contact site only when membranes are closely apposed, or local proteins that shift after drug treatment, mutation, or stimulation.
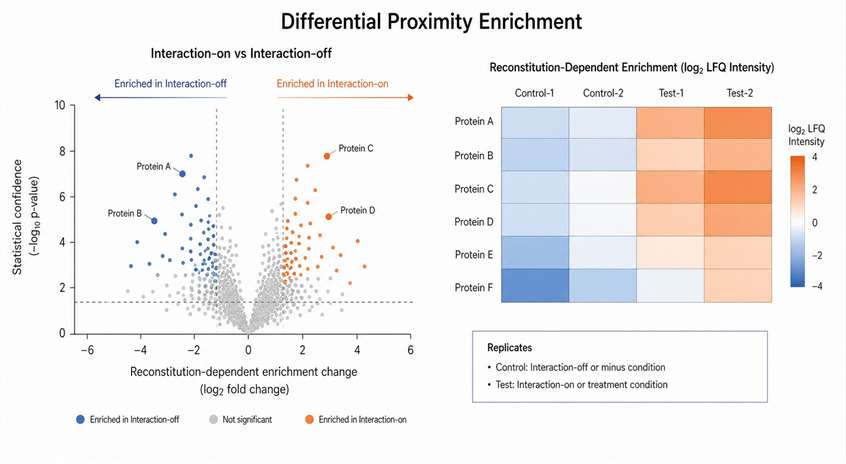
We interpret split proximity labeling data with care. The LC-MS/MS output represents a reconstitution-dependent proximal proteome. It can identify proteins near the interaction or contact event, but it does not prove that every detected protein directly binds the bait or prey. We help you plan controls and background filtering so the dataset supports candidate prioritization and follow-up validation.
What Split Proximity Labeling Detects
Split-TurboID or Split-APEX proteomics can identify:
- Proteins enriched near a defined protein-pair interaction
- Proteins associated with organelle or membrane contact sites
- Local proteins that appear when a signaling complex forms
- Proteins that change proximity after drug treatment or stimulation
- Candidate regulators near interaction-dependent microenvironments
- Contact-site or condition-dependent proximal protein networks
Why Split Enzymes Add Specificity
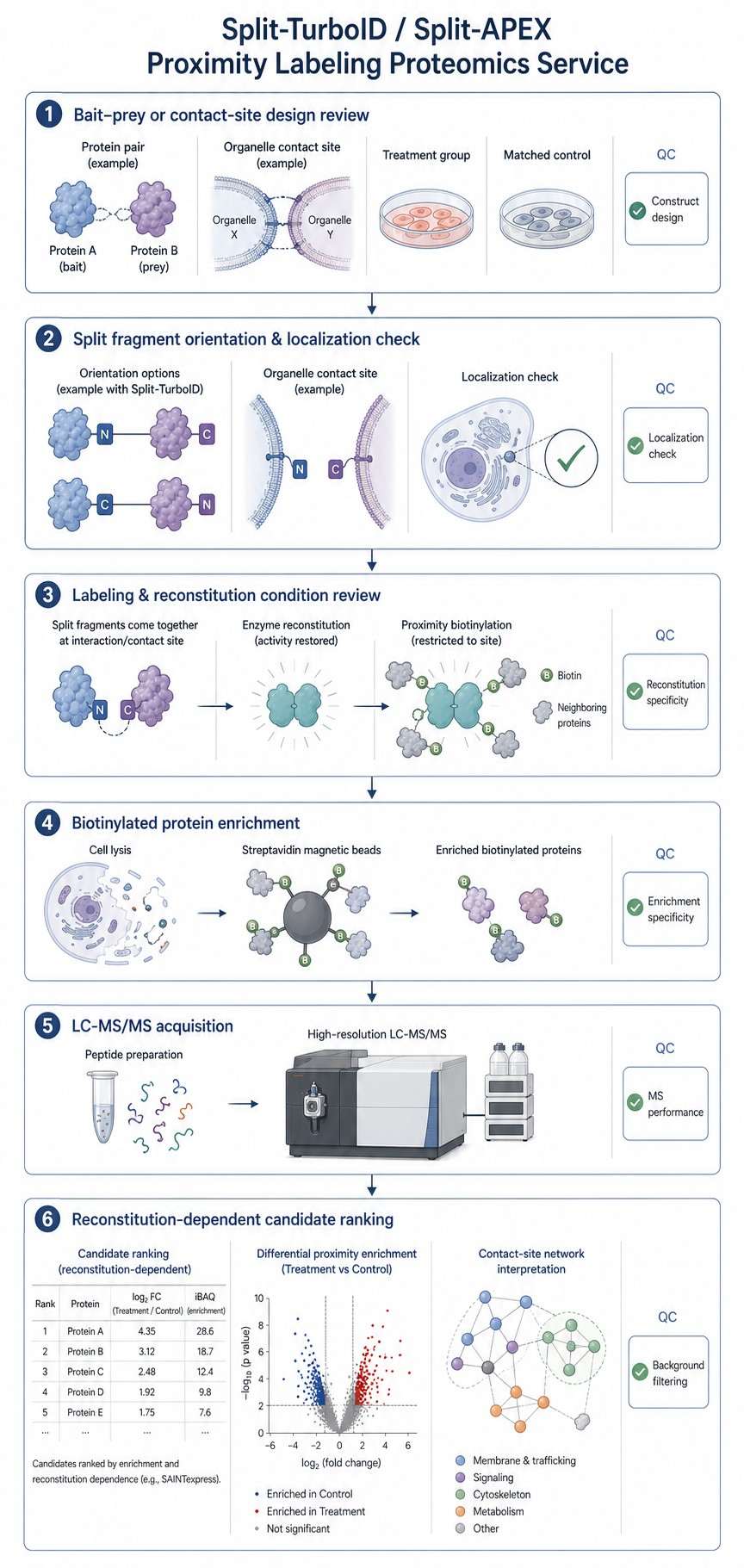
Full-length APEX, TurboID, or BioID can label proteins near a bait or compartment even when a specific interaction is not occurring. Split systems add another layer of control because labeling depends on fragment proximity and enzyme reconstitution.
This does not mean background disappears. Fragment orientation, expression level, localization, linker design, and controls all affect specificity. A well-designed split proximity labeling project should include controls that test whether labeling is truly driven by the intended interaction or contact event.
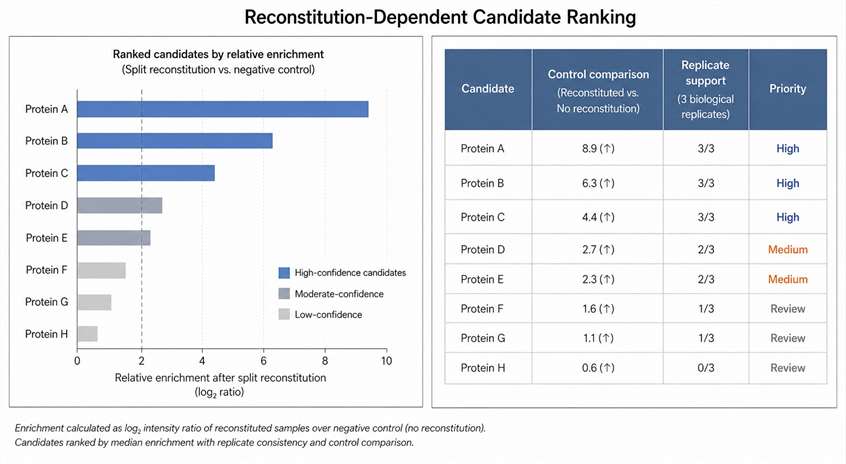
How to Interpret Reconstitution-Dependent Evidence
A strong candidate is usually supported by enrichment over control, consistency across replicates, and biological relevance to the protein pair, contact site, treatment condition, or pathway state. For direct-binding or functional conclusions, top candidates should be validated by orthogonal methods such as Co-IP, imaging, mutagenesis, targeted MS, functional perturbation, or structural MS.