Luo S., Xie L., Yang L., Hu Z., Wang L., Wang Y., Li Q., Guo S., Tao S., Jiang H. "Sensitive and specific affinity purification-mass spectrometry assisted by PafA-mediated proximity labeling." Cell Reports Methods 2025;5:101166. https://doi.org/10.1016/j.crmeth.2025.101166
Scientific Challenge
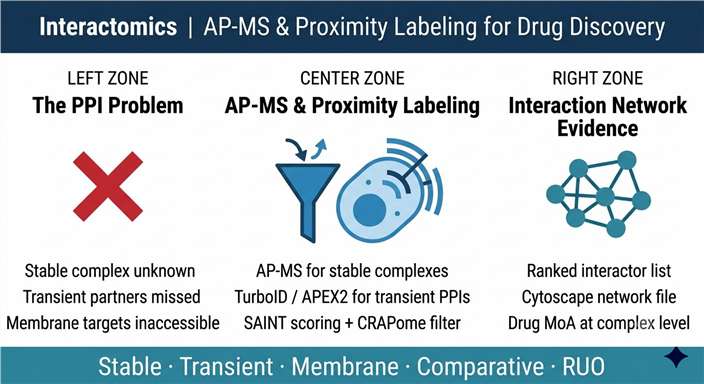
AP-MS is the workhorse of protein interactomics, but two persistent limitations constrain its use in drug discovery: high non-specific binding that inflates false-positive interaction lists, and the inability to capture weak, transient, or cell-surface interactions that are lost during cell lysis and stringent washing. Membrane receptor interactomics — mapping the signalling complex around a GPCR, for example — is particularly affected because detergent extraction disrupts the native membrane context that governs receptor–effector recruitment. The researchers at Shanghai Jiao Tong University developed APPLE-MS (Affinity Purification coupled Proximity LabEling–Mass Spectrometry), combining the high specificity of Twin-Strep-tag enrichment with PafA-mediated proximity labeling, to address these limitations simultaneously.
Methods
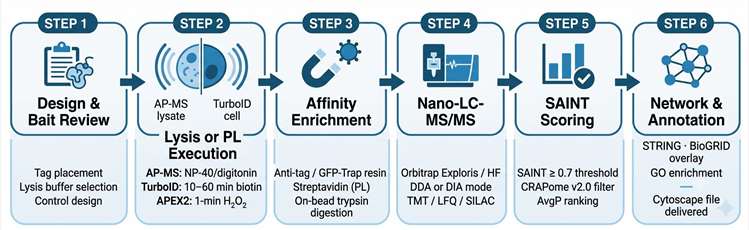
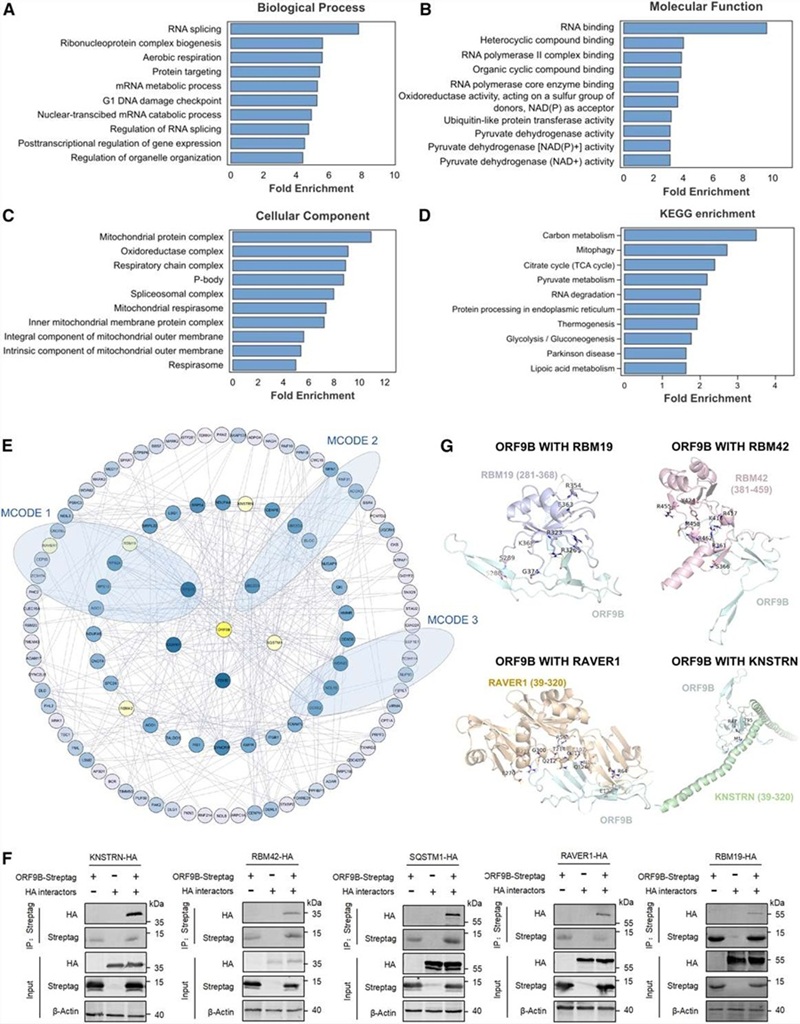
APPLE-MS was benchmarked against standard AP-MS across three biological targets in HEK293T and INS-1E cells. Twin-Strep-tag enrichment provides the affinity purification specificity layer, while PafA-mediated covalent biotinylation of proximal proteins captures interactions within the native cellular environment. The method was applied to: (1) SARS-CoV-2 ORF9B, profiling its dynamic mitochondrial interactome during antiviral response across multiple time points; (2) endogenous PIN1, a prolyl-isomerase implicated in signalling regulation, using APPLE-MS versus standard AP-MS side-by-side; and (3) GLP-1 receptor (GLP-1R), a drug target of major therapeutic interest, enabling in situ mapping of its membrane-proximal complex without cell lysis.
Key Results
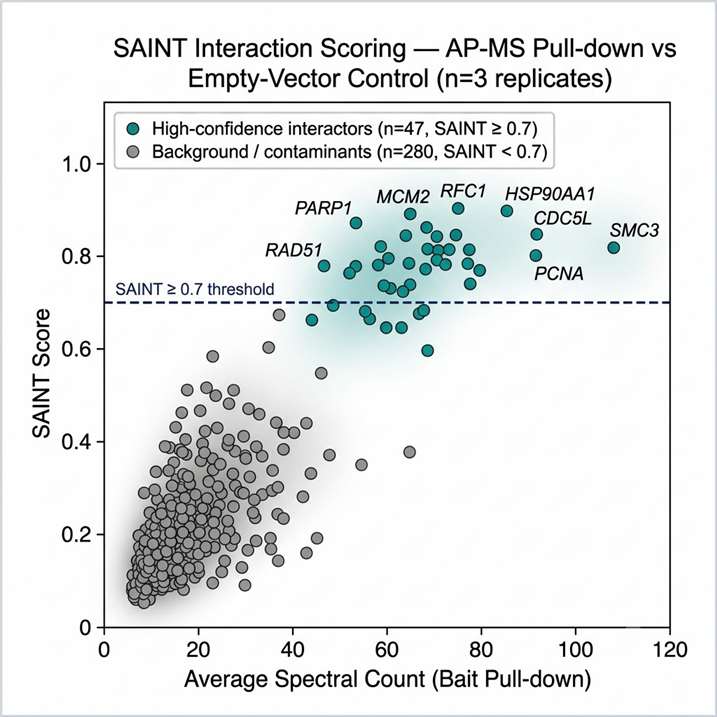
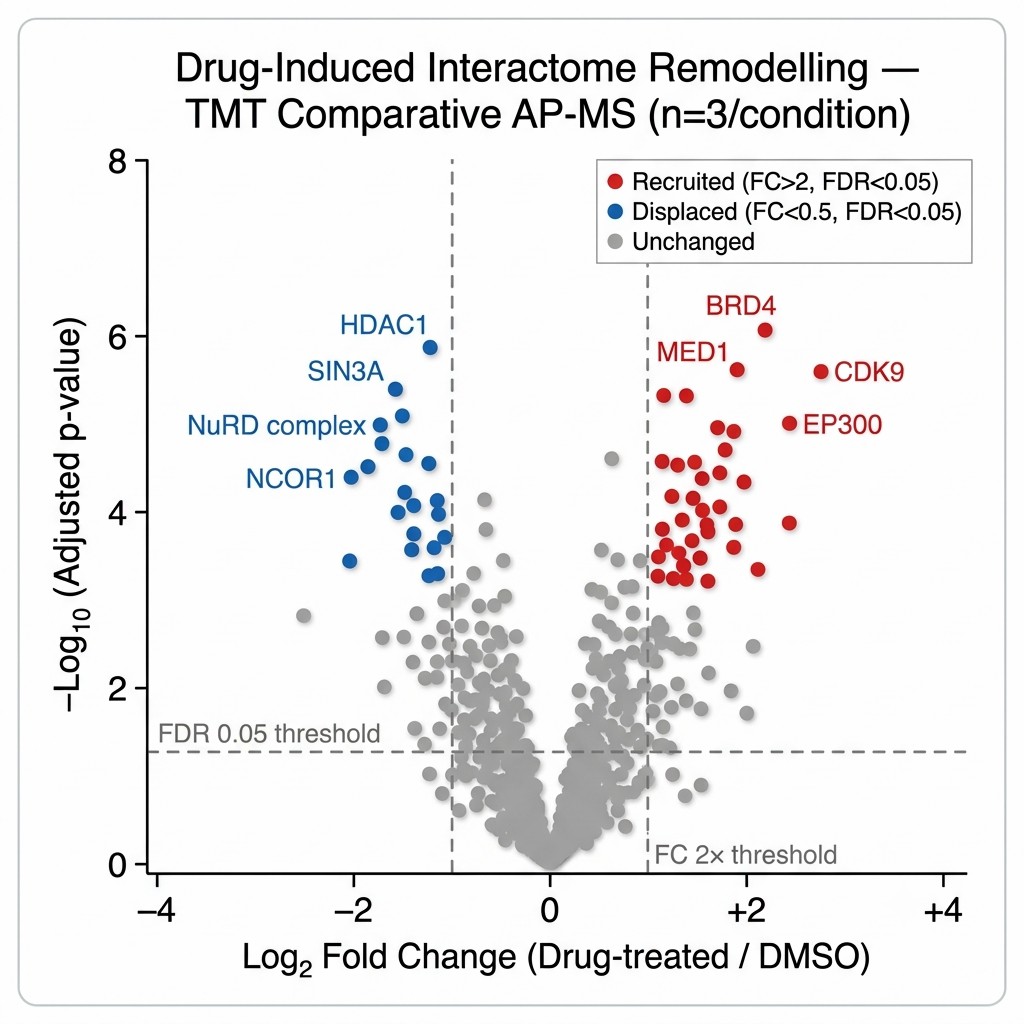
APPLE-MS achieved a 4.07-fold improvement in specificity over standard AP-MS, as measured by the ratio of true interactors (database-validated) to total identified proteins. For SARS-CoV-2 ORF9B, the method revealed a time-resolved mitochondrial interactome during poly(I:C)-induced antiviral responses, identifying dynamic interactions with metabolic regulators (including TUFM) and immune modulators (including RNF123) — providing mechanistic insight into how ORF9B reprogrammes host metabolism during infection. For endogenous PIN1, APPLE-MS uncovered novel roles in DNA replication stress responses and RNA processing, identifying associations with the MCM complex and RFC1 that extend PIN1's known function beyond canonical prolyl-isomerase activity. Critically, APPLE-MS enabled in situ mapping of GLP-1R membrane complexes in HEK293T and INS-1E cells — identifying canonical and previously uncharacterised co-receptors and signalling components — demonstrating that the method can access interaction networks at the cell surface that are invisible to conventional AP-MS.
Significance for Interactomics Drug Discovery Services
This study demonstrates three capabilities directly relevant to pharmaceutical interactomics: the specificity improvement addresses the false-positive problem that has historically limited AP-MS utility in lead optimisation; the GLP-1R application demonstrates that membrane receptor signalling complexes — a major class of drug targets — are now mappable by proximity-labeling interactomics; and the time-resolved ORF9B profiling shows that dynamic, drug-relevant interaction changes can be captured with temporal resolution. Figure 2 of the paper directly compares APPLE-MS versus standard AP-MS specificity, providing a quantitative benchmark for the method's performance advantage.