Integrated LiP-MS + Metabolomics for Target Deconvolution and MoA Elucidation
Integrated target deconvolution and mechanism-of-action profiling from a single biological experiment.
Phenotypic screening remains one of the most powerful strategies for discovering first-in-class therapeutics, yet it presents a fundamental challenge: a compound shows clear bioactivity, but its molecular target and mechanism of action remain unknown. Resolving this question has traditionally required running separate, disconnected assays — one for target identification and another for pathway-level readout — often on different platforms, with different samples, and at different times.
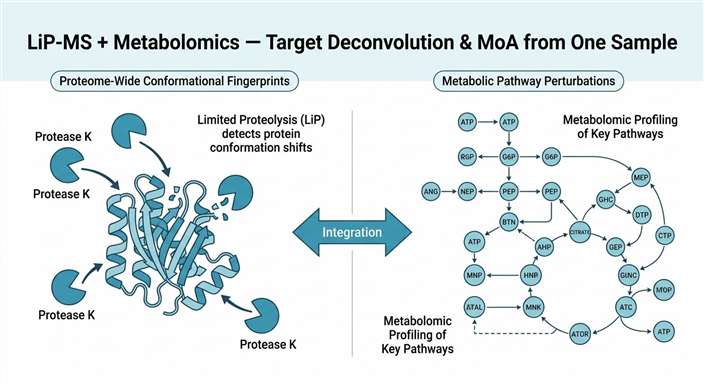
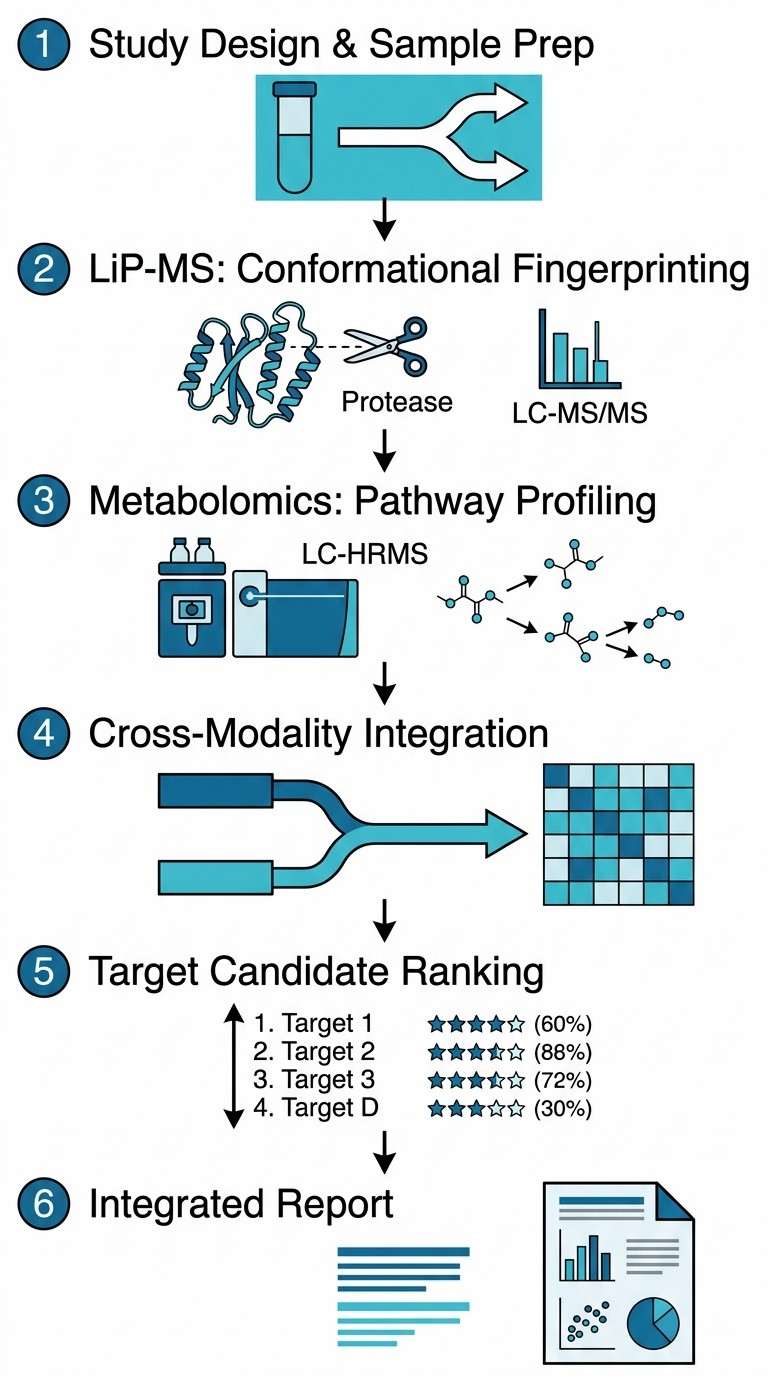
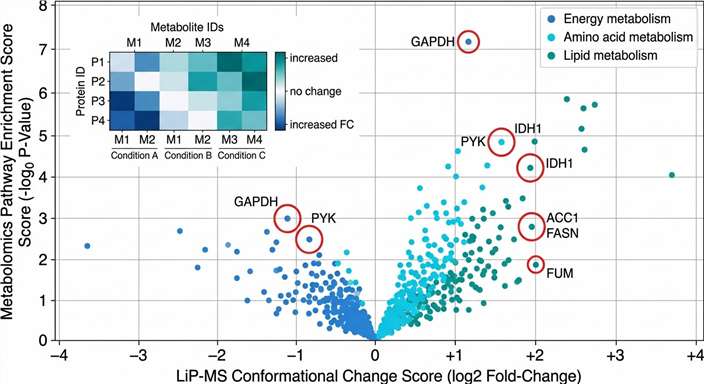
We designed our combined LiP-MS + Metabolomics service to close this gap. By running Limited Proteolysis–Mass Spectrometry (LiP-MS) and untargeted metabolomics on the same biological samples, we deliver two orthogonal layers of evidence from a single experiment. LiP-MS detects drug- or metabolite-induced conformational changes across the proteome, pinpointing the proteins that directly engage with your compound. Metabolomics captures the resulting shifts in metabolite abundance, revealing which metabolic pathways are activated, inhibited, or rewired as a consequence of target engagement.
Together, these two data types answer the question that neither can answer alone: not just which protein your compound binds, but what happens next. For discovery teams facing phenotypic hits with unknown targets, this integrated approach reduces the time from hit to mechanism hypothesis, eliminates the risk of cross-platform sample variability, and provides a single, coherent data package ready for internal review or publication.
Our service is part of the broader multi-omics integration platform at Creative Proteomics, and complements our standalone LiP-MS service by adding the metabolomics dimension that connects target engagement to functional outcome.