Mechanism-of-Action (MoA) Analysis Service — From Phenotypic Activity to Confirmed Mechanism
You have a compound that works. The question is how.
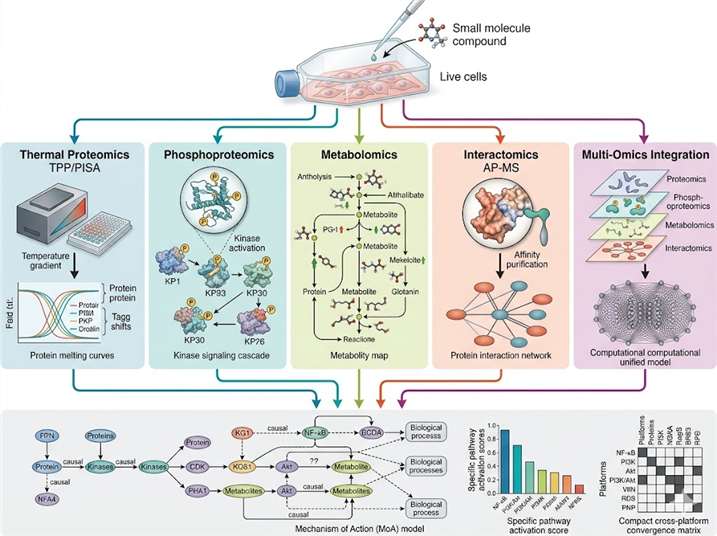
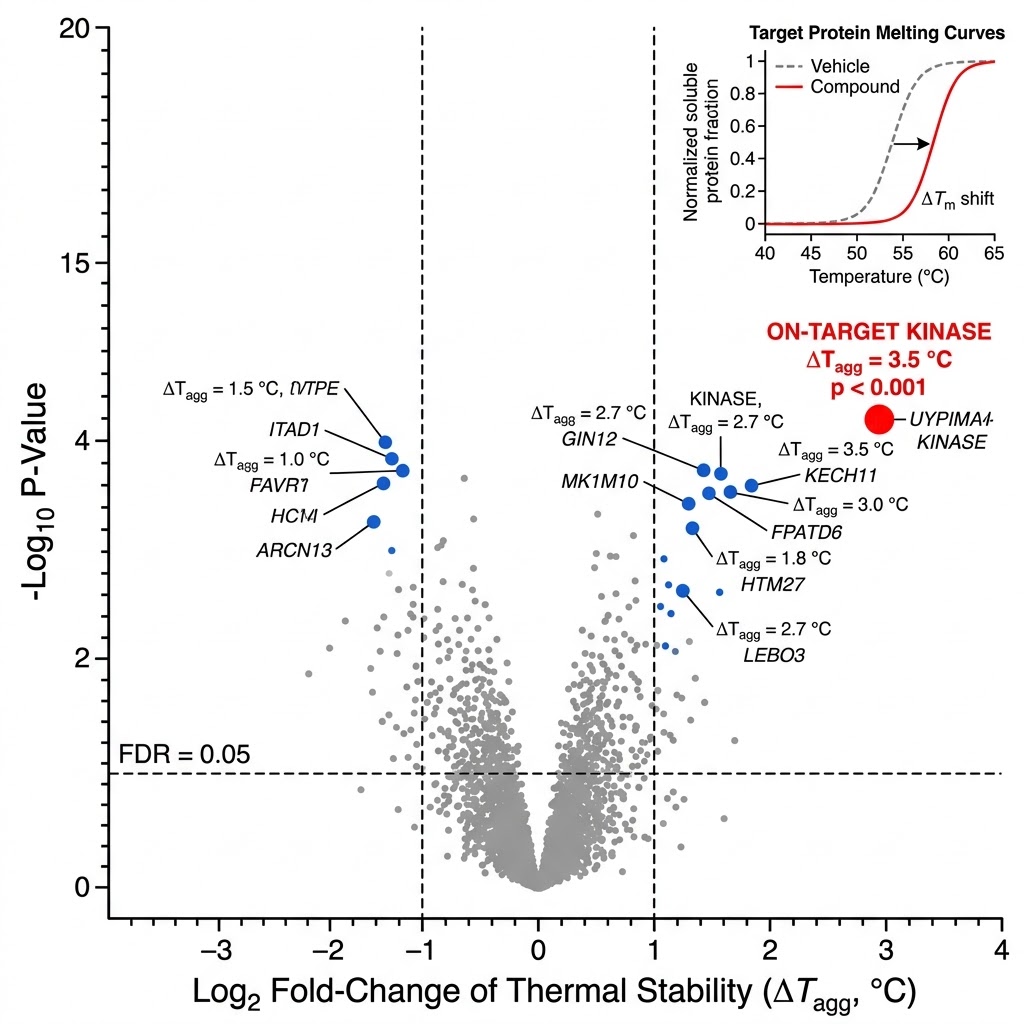
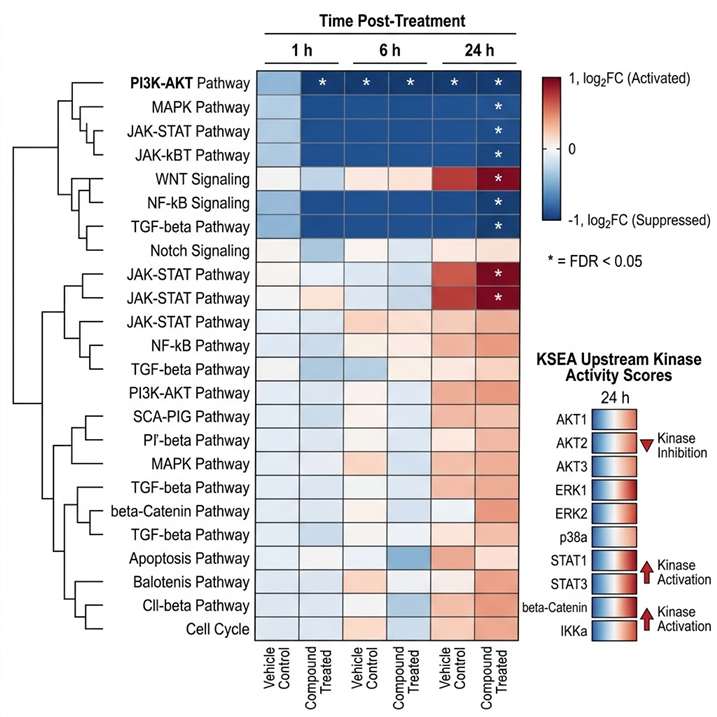
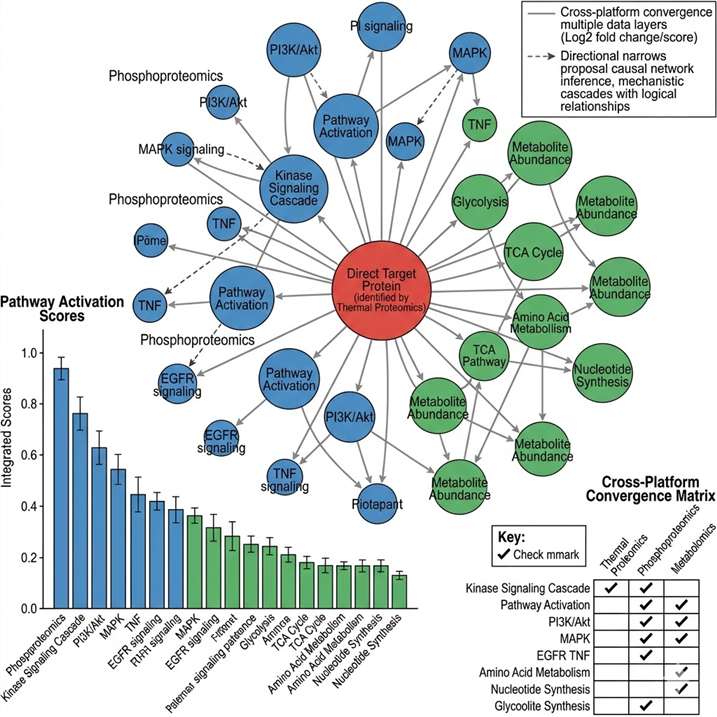
Target identification tells you which protein your compound binds. Target engagement tells you whether it binds in cells. MoA analysis goes further: it reveals the downstream consequences of that binding — which signaling cascades are activated or suppressed, which metabolic pathways shift, how protein interaction networks rewire, and ultimately, how the cellular phenotype emerges from the molecular interaction.
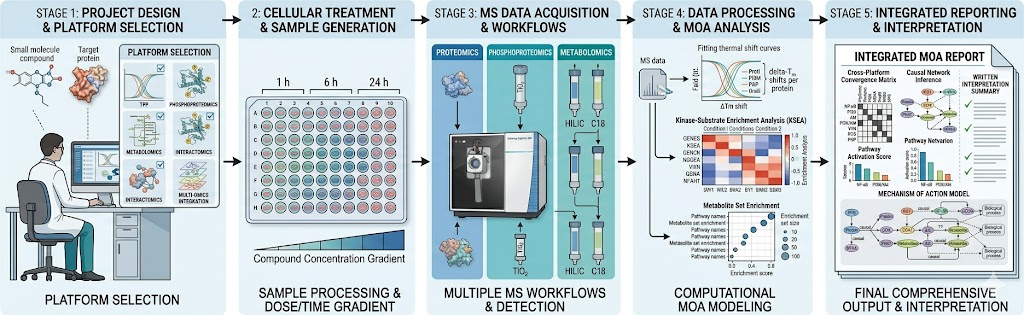
At Creative Proteomics MassTarget, we deploy an integrated multi-omics MoA analysis platform combining thermal proteomics, phosphoproteomics, metabolomics, interactomics, and multi-omics integration to deliver a complete mechanistic picture. Our service is built for drug discovery teams who need to understand not just what their compound engages, but how that engagement produces the observed biology. For dedicated target identification workflows, our thermal proteomics for MoA service provides the first critical layer of mechanistic evidence.
Key Advantages:
- Multi-platform MoA elucidation — proteomics + phosphoproteomics + metabolomics under one project.
- Quantitative pathway-level readouts with statistical confidence.
- Cellular context preserved throughout — no buffer artefacts.
- Orthogonal cross-platform validation built into every MoA study.
- Low compound consumption: 2–10 mg sufficient for comprehensive multi-omics MoA.
- Turnaround: 3–8 weeks depending on platform scope.