Structure–Activity Relationship by Mass Spectrometry (SAR-MS) Service
Direct binding measurement for SAR-driven lead optimization.
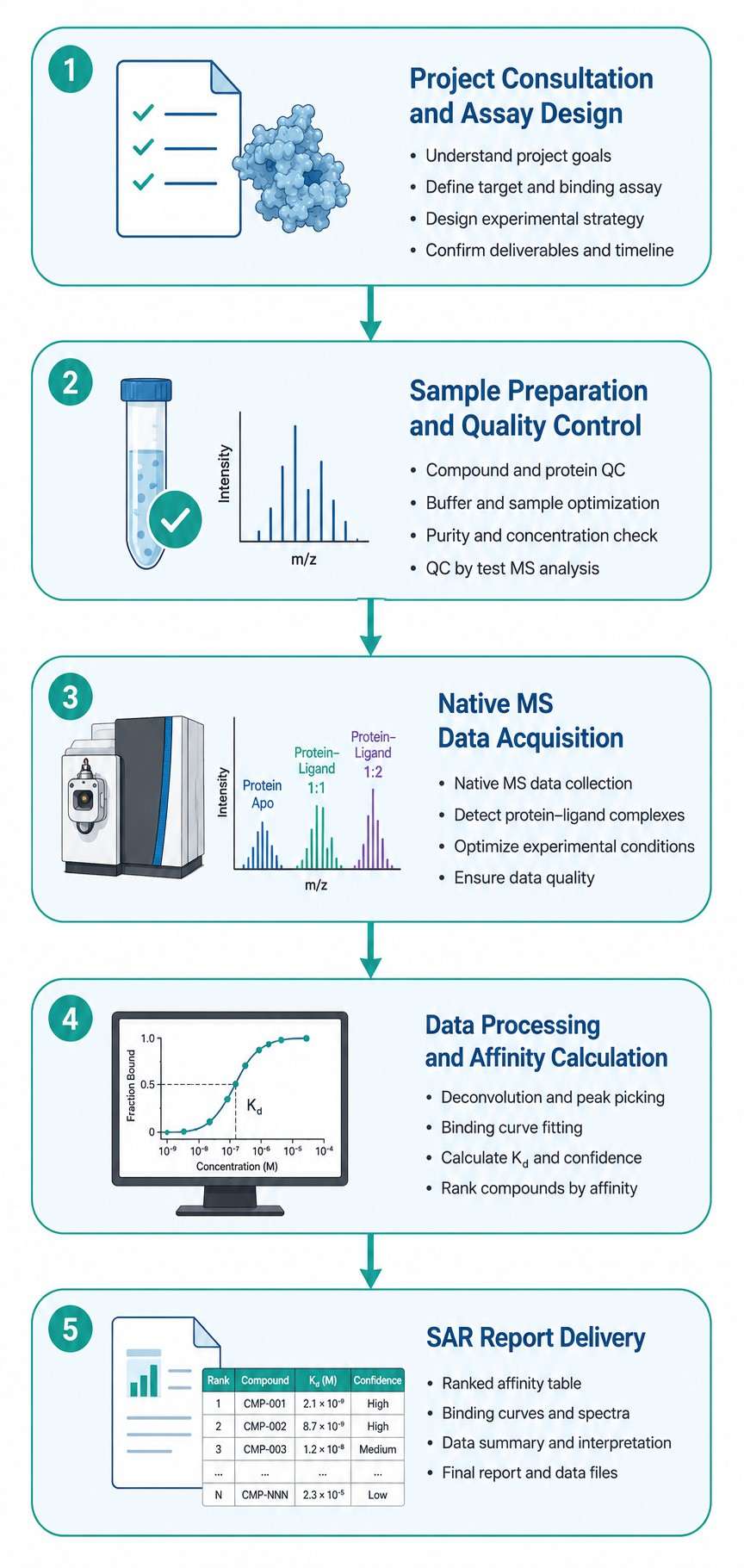
Every analog synthesized needs to be tested for binding — and the quality of that data determines how efficiently your team can optimize potency, selectivity, and drug-like properties. Our SAR-MS service delivers direct biophysical binding affinity measurements by native mass spectrometry, without labels, immobilization, or computational modeling.
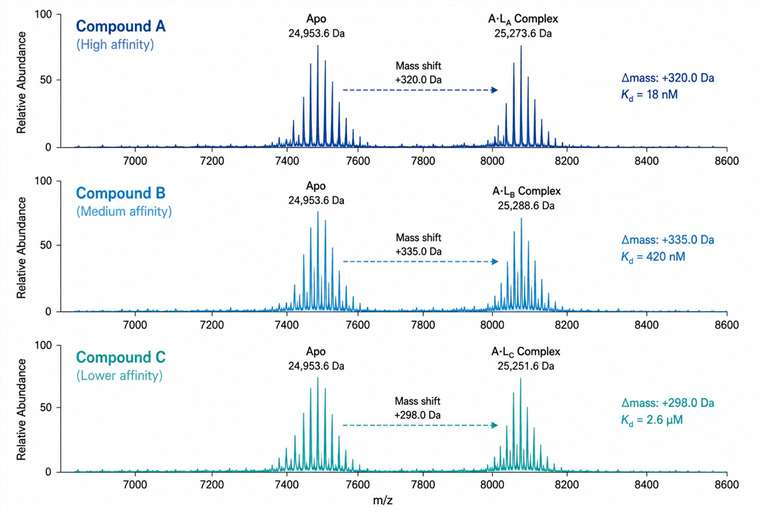
We measure protein–ligand binding under native solution conditions across a broad affinity range (nM to mM), providing Kd values, binding stoichiometry, and rank-ordered affinity lists for your chemical series. Whether you are optimizing a fragment hit, profiling a congeneric series, or assessing selectivity across related targets, SAR-MS gives you the data you need to make confident SAR decisions.
Key Advantages:
- Label-free direct binding — no fluorescent tags, radioactive labels, or immobilization artifacts
- Broad affinity range — detects binders from nM potent leads to mM weak fragments
- Compound rank ordering — clear affinity ranking within chemical series
- Multi-target selectivity — same platform works across diverse protein targets
- Native solution conditions — measurements in physiological-like buffers
- Rapid turnaround — results in days, not weeks