What is SPROX-MS? (Stability from Rates of Oxidation)
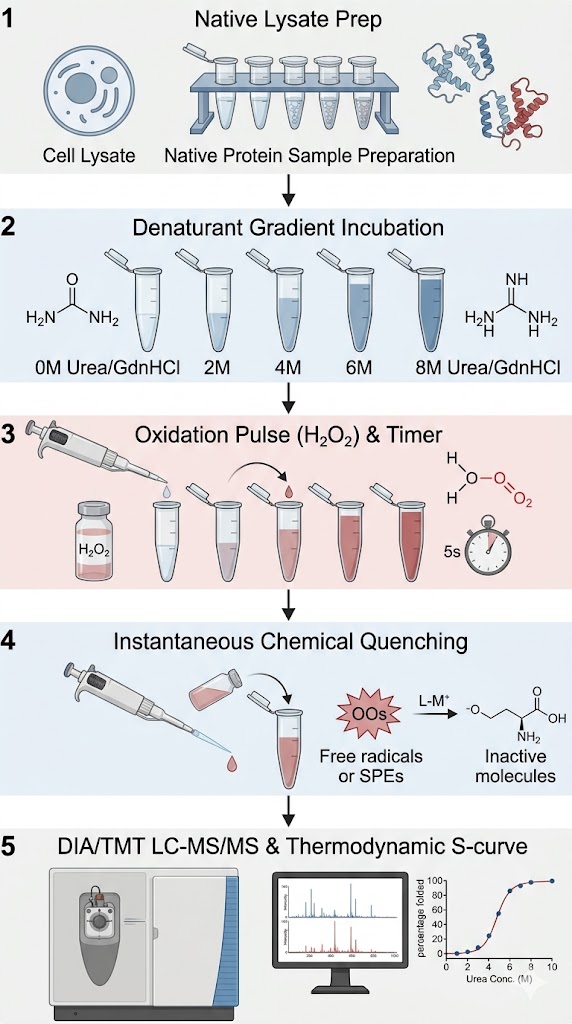
Stability of Proteins from Rates of Oxidation (SPROX) coupled with Mass Spectrometry (MS) is an advanced, label-free chemoproteomic technology designed to measure the thermodynamic stability of proteins and detect precise ligand-binding interactions. Unlike traditional assays that rely on physical heating, SPROX utilizes chemical denaturants and a highly specific oxidation reaction to probe protein structures directly within complex biological mixtures.
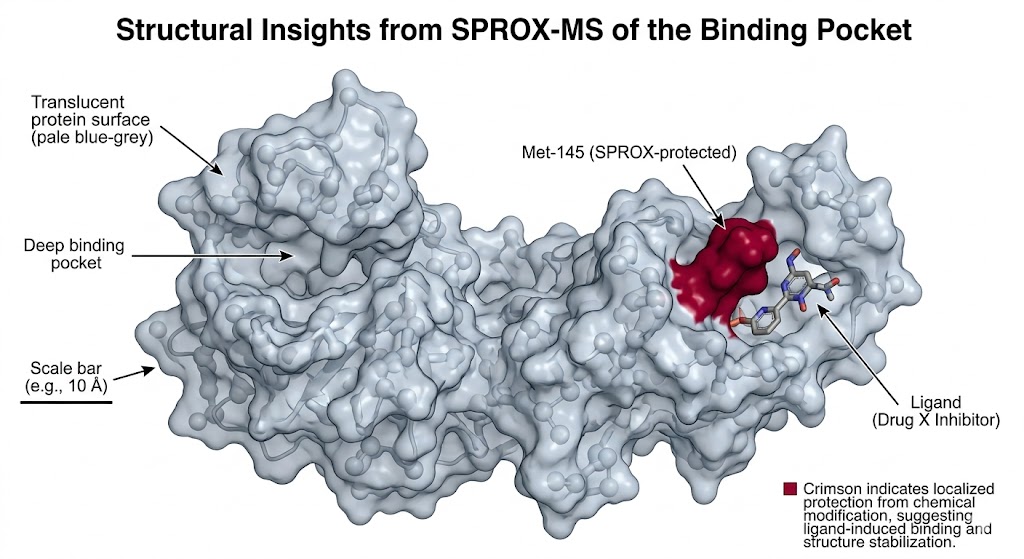
The biophysical principle behind SPROX-MS is elegantly straightforward yet incredibly powerful. In a folded, native protein, hydrophobic amino acids—particularly Methionine residues—are typically buried deep within the structural core or shielded at the protein-protein interaction interfaces. Because they are hidden, they are protected from reacting with the surrounding solvent. However, when we introduce a chemical denaturant like urea or guanidine hydrochloride (GdnHCl), the protein begins to unfold. As the protein unfolds, these previously hidden Methionine residues become exposed to the solvent.
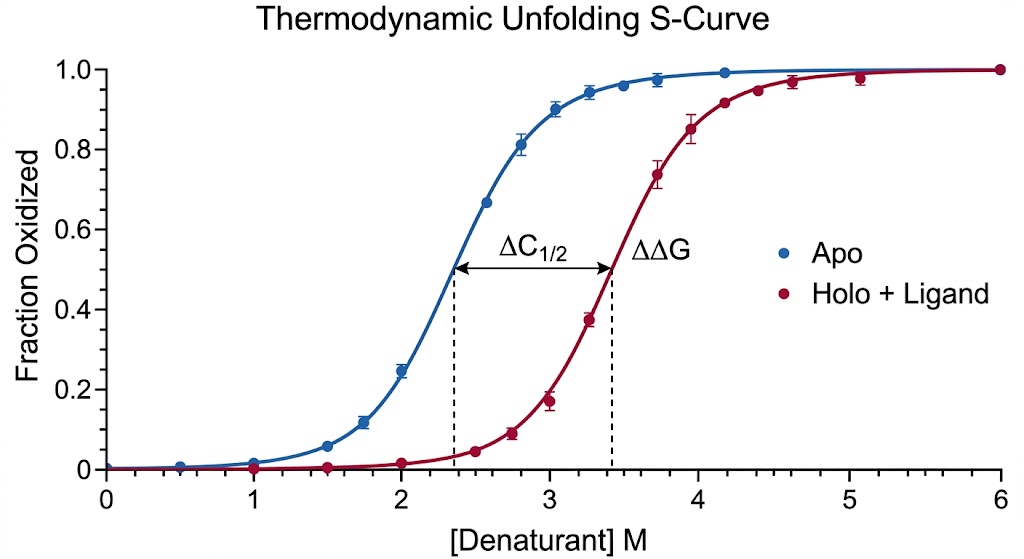
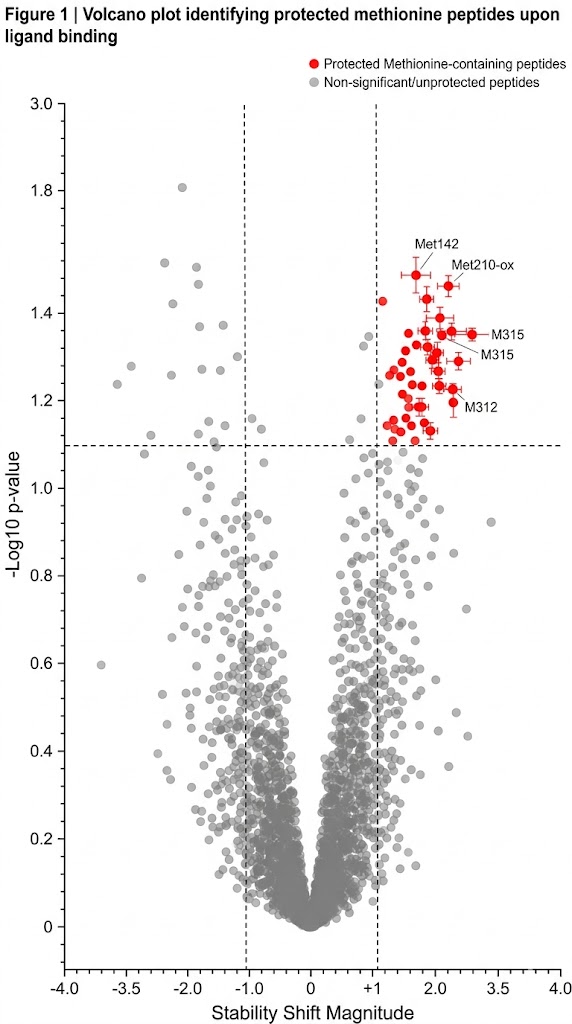
During the SPROX assay, we introduce a brief, highly controlled pulse of hydrogen peroxide (H2O2). The exposed Methionine residues rapidly react with the hydrogen peroxide, undergoing a covalent oxidation event that adds exactly one oxygen atom (+16 Da mass shift) to the amino acid side chain, creating methionine sulfoxide. By utilizing high-resolution mass spectrometry, we can quantify the exact ratio of oxidized versus unoxidized methionine for thousands of peptides simultaneously. When a small molecule drug binds to a protein, it inherently stabilizes the folded state, making it much harder for the chemical denaturant to unfold the protein. This stabilization delays the exposure of the Methionine residue, resulting in a measurable shift in the oxidation rate. By tracking this shift, we identify exactly which proteins your drug bound to, and precisely how tightly it held on.