Hydroxyl Radical Protein Footprinting: A Mass Spectrometry-Based Structural Method for Studying the Higher Order Structure of Proteins. https://pubs.acs.org/doi/10.1021/acs.chemrev.1c00432
Background
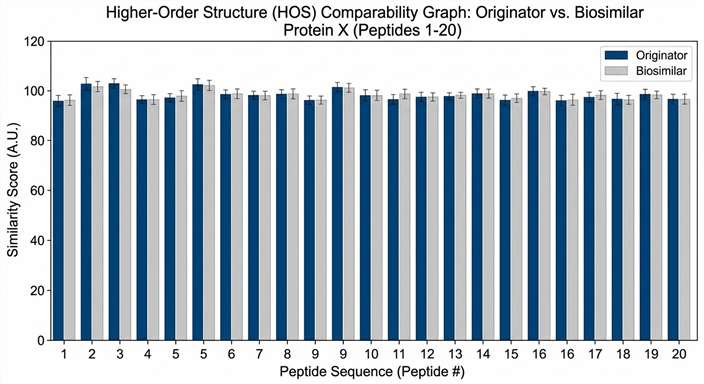
Understanding the higher-order structure (HOS) of proteins and their biological complexes is vital for developing safe and effective biotherapeutics. Traditional structural biology methods like X-ray crystallography often struggle with highly flexible proteins or force the molecules into rigid, unnatural crystal lattices that do not reflect their true dynamic behavior in the human body. Furthermore, evaluating the structural comparability of biosimilars requires robust methods that can sensitively detect minute conformational deviations in solution. Researchers required a powerful analytical method to map solvent-accessible surfaces in a native liquid environment without suffering from the back-exchange limitations of standard techniques.
Methods
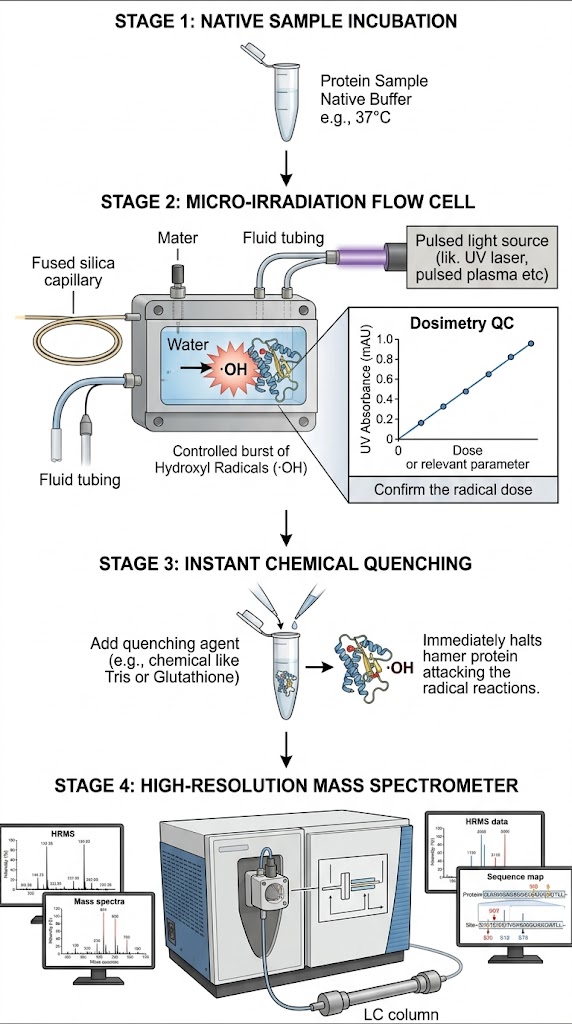
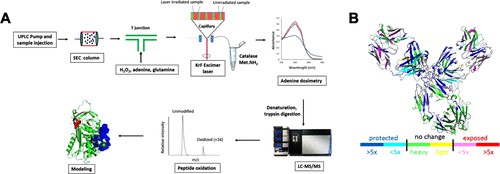
Researchers utilized Hydroxyl Radical Protein Footprinting (HRF-MS) to accurately probe the structural topography of therapeutic target proteins in their native solution state. By exposing the proteins to a carefully controlled, brief burst of hydroxyl radicals, they irreversibly labeled the solvent-exposed amino acids. Crucially, the radical dosage was strictly monitored in real-time using an internal dosimeter to prevent protein unfolding or biologically irrelevant over-oxidation. The stably labeled samples were then subjected to thorough, aggressive enzymatic digestion—which would have easily destroyed a traditional reversible HDX label—and analyzed using high-resolution liquid chromatography-tandem mass spectrometry (LC-MS/MS) to identify and quantify the exact sites of oxidation.
Results
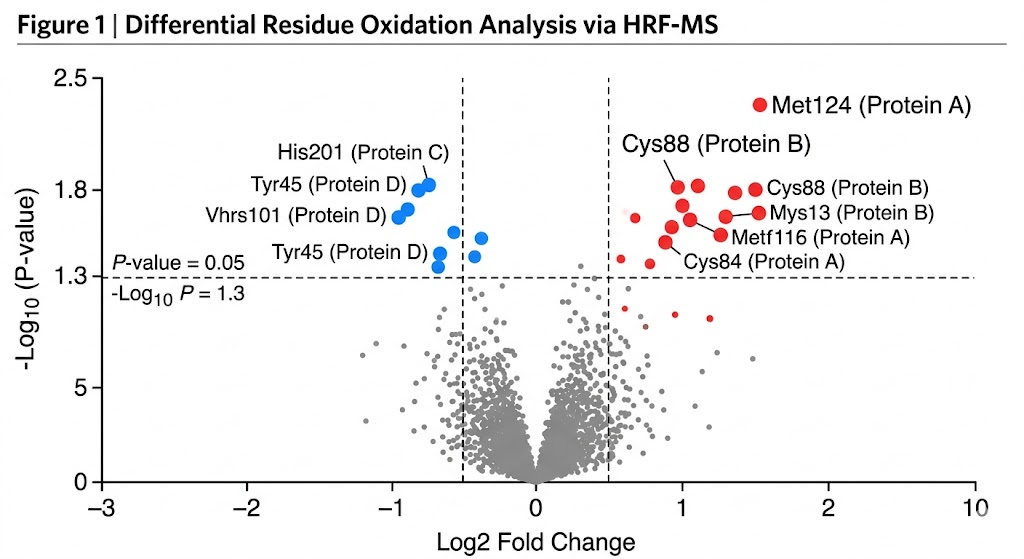
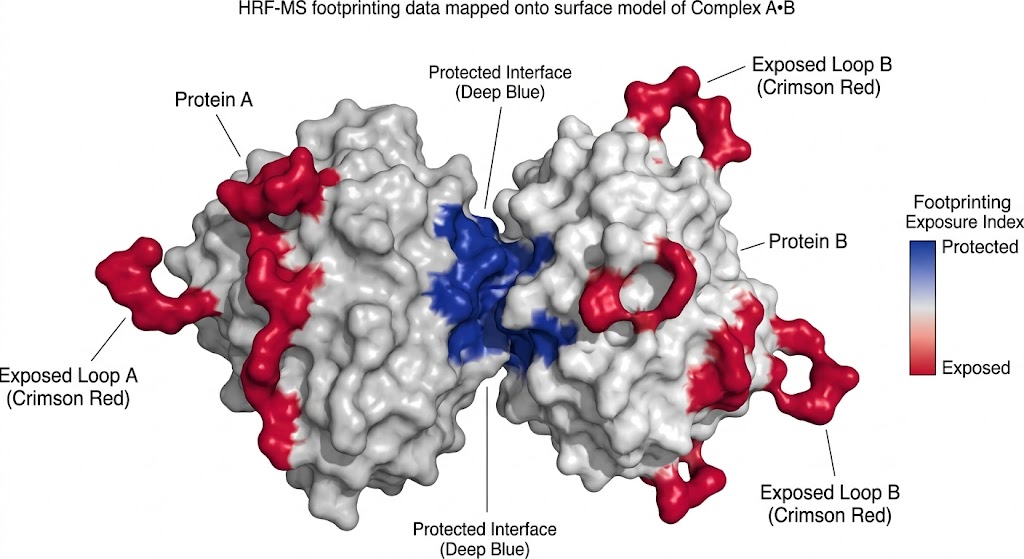
As detailed in the comprehensive footprinting data and structural mapping models, HRF-MS successfully mapped the complex solvent-accessible surfaces of the target proteins. The mass spectrometry data accurately quantified specific, localized oxidation events—such as stable +16 Da and +32 Da oxygen additions to exposed aromatic residues (tyrosine, tryptophan, phenylalanine) and sulfur-containing residues (methionine). This massive, high-confidence dataset allowed the researchers to build high-fidelity, residue-level topographical maps that precisely pinpointed critical interaction interfaces and revealed subtle conformational changes that other analytical methods completely missed.
Conclusion
The extensive review demonstrates that HRF-MS is an exceptionally robust, mass spectrometry-based structural method. Its unique irreversible covalent labeling mechanism makes it uniquely powerful for evaluating the higher-order structure of challenging protein therapeutics, surviving rigorous downstream processing, and securing the critical, high-resolution structural data necessary for advanced drug development and rigorous regulatory evaluation by agencies worldwide.