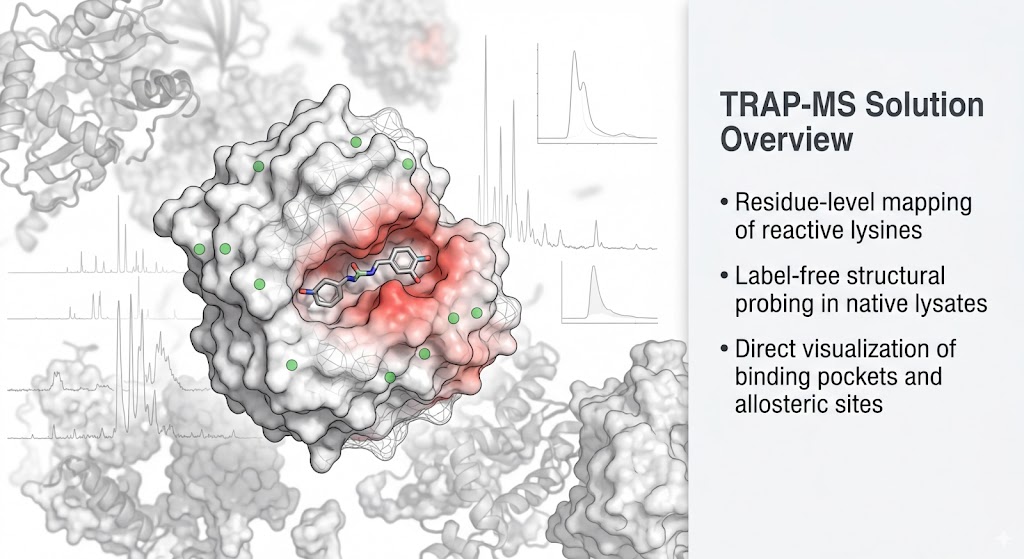
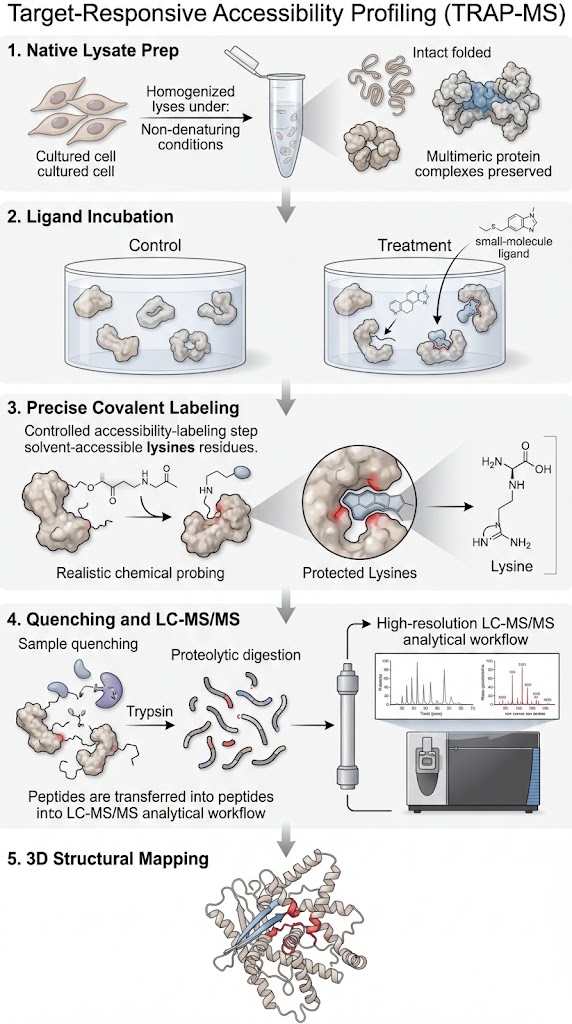
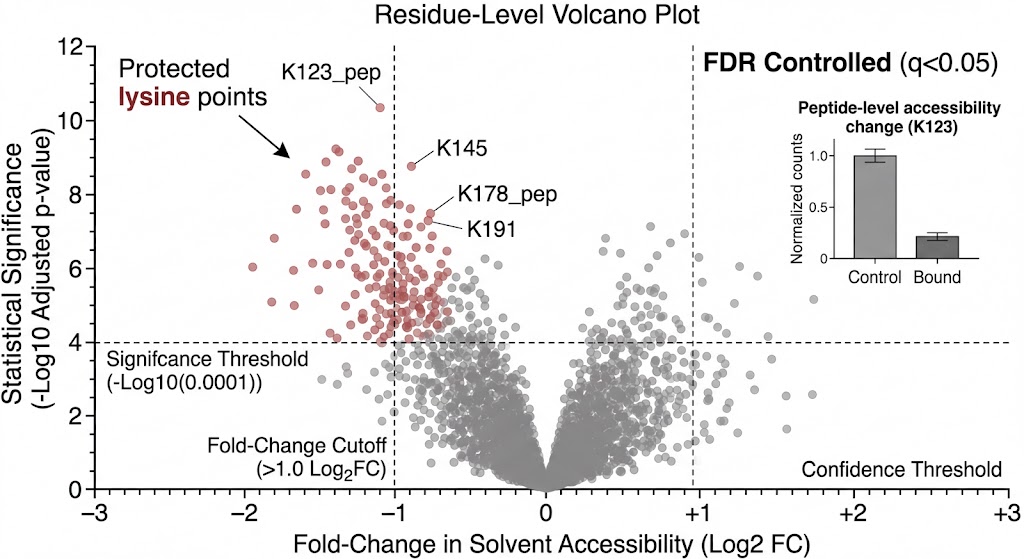
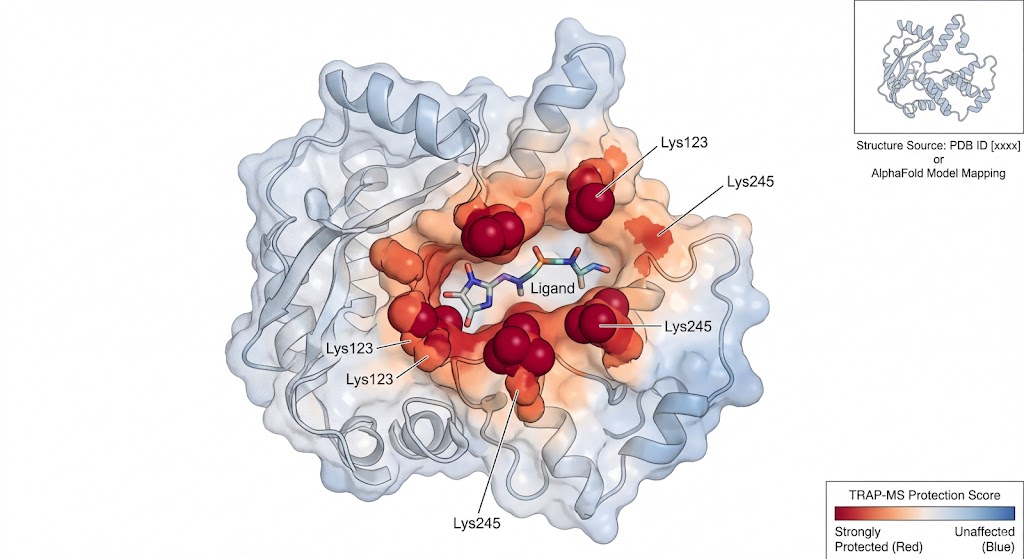
Why TRAP? The Bridge Between Target ID and Structural Biology
For decades, drug discovery has faced a frustrating gap. Traditional target deconvolution methods can tell you which protein your drug binds to, but they leave medicinal chemists blind as to where it binds. Without knowing the 3D structure of the binding pocket, optimizing the drug's affinity and selectivity is incredibly difficult.
Historically, researchers turned to X-ray crystallography or Cryo-EM to get this structural data, but these methods require heavily purified proteins and fail frequently. Another alternative is HDX-MS / HDX-driven Epitope Mapping, which offers fantastic peptide-level resolution. However, HDX-MS also strictly requires highly purified, recombinant protein. If your target is a complex membrane protein, an intrinsically disordered protein (IDP), or a target that loses its natural conformation outside of the cell, HDX-MS simply cannot be performed.
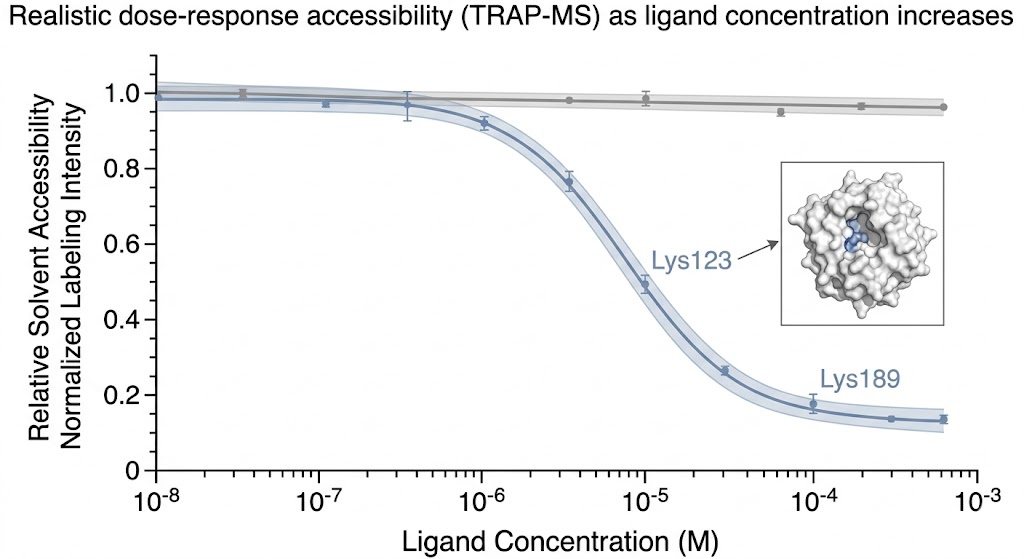
This is exactly why we utilize TRAP-MS. TRAP operates perfectly in native cell lysates and tissue homogenates. It allows us to probe the structural accessibility of proteins while they remain in their natural, physiological environment, surrounded by their native interacting partners. It bridges the gap by providing high-resolution structural mapping without the impossible prerequisite of protein purification.