Background
Type I interferon signaling is controlled by dynamic protein assemblies. These assemblies can change after stimulation and may include weak, transient, or context-dependent associations. Conventional interaction methods may miss part of this landscape because the signaling state can be altered during extraction or purification.
Schiefer and Hale used TurboID-based proximity labeling to map the proximal proteomes of seven canonical type I interferon signaling cascade members under basal and IFN-stimulated conditions. The study focused on IFNAR1, IFNAR2, JAK1, TYK2, STAT1, STAT2, and IRF9, covering receptor-level, kinase-level, transcription-factor-level, and regulatory pathway components.
Methods
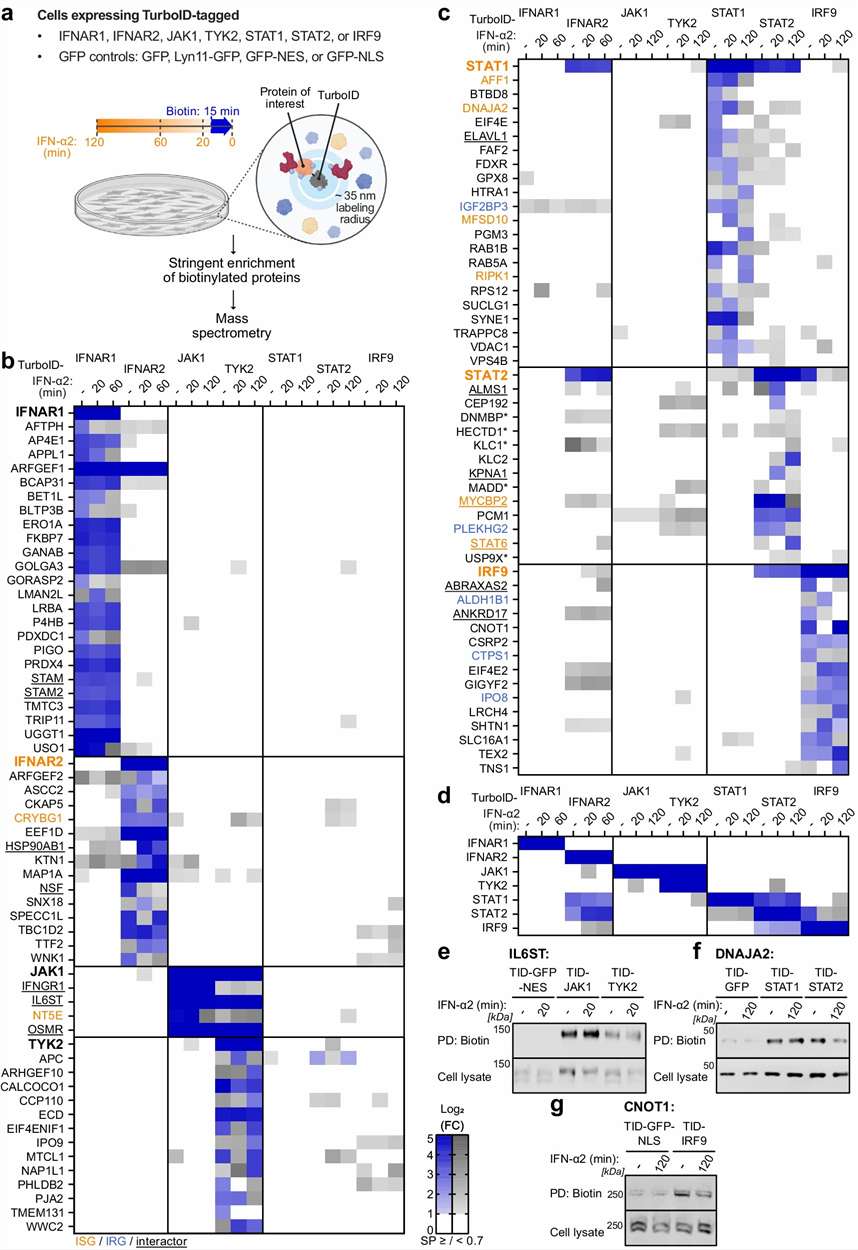
The researchers generated cell systems expressing TurboID-tagged IFN signaling proteins. Cells were either left under basal conditions or stimulated with IFN-α2. Before harvest, biotin was added so that TurboID could label nearby proteins in the cellular environment.
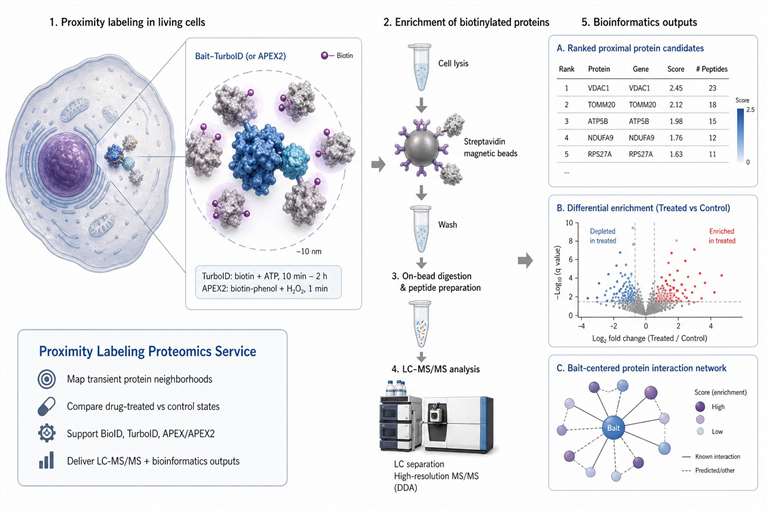
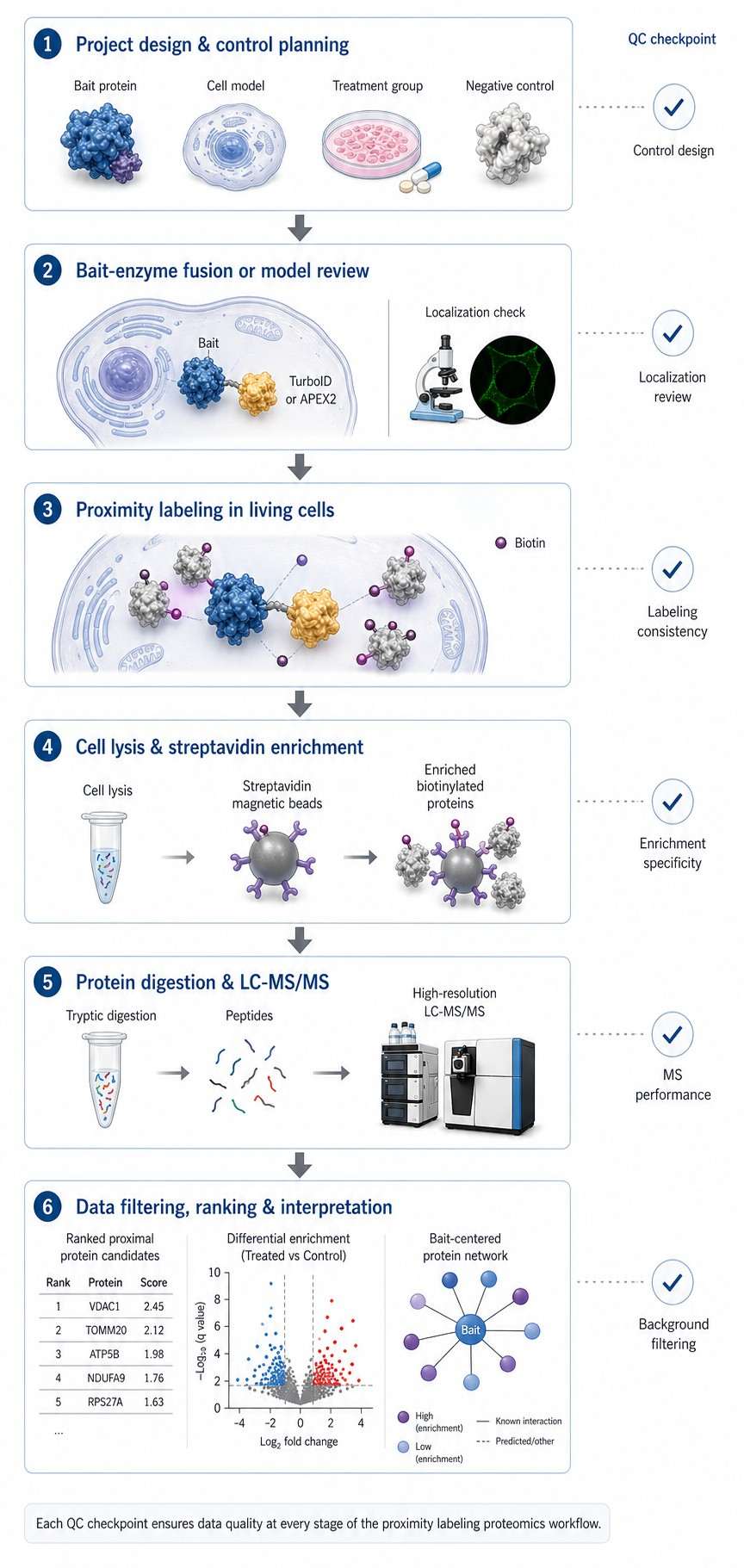
After labeling, biotinylated proteins were enriched and analyzed by label-free mass spectrometry. The study then compared proximal protein profiles across signaling components and stimulation states. Figure 1 presents the system-wide strategy, including TurboID tagging, biotin labeling, enrichment, label-free MS analysis, heatmap visualization, and validation of selected proteins.
Results
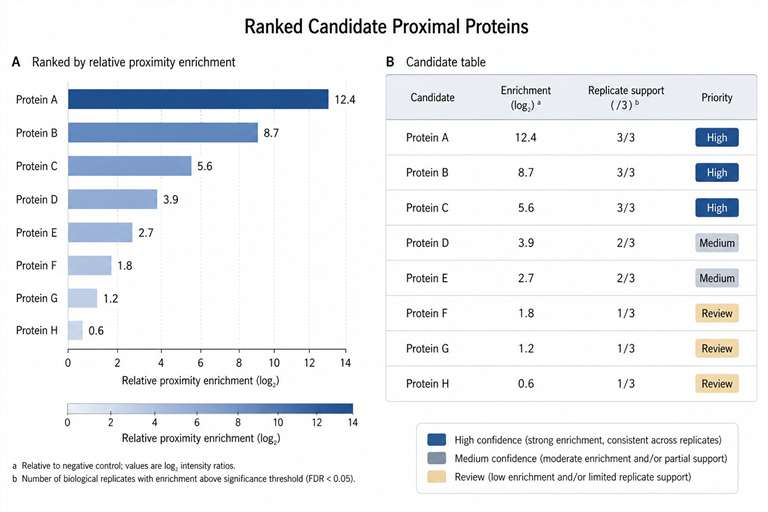
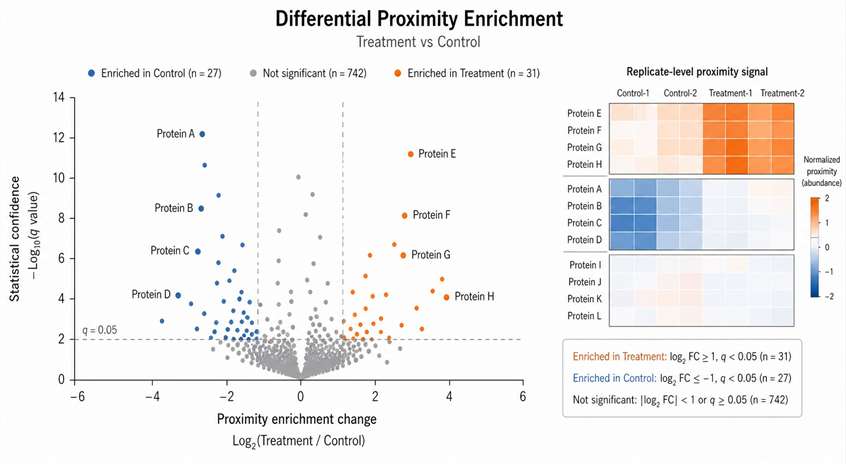
The study identified 103 high-confidence proteins proximal to type I interferon signaling components. The dataset included known pathway-associated proteins as well as additional candidates that were not previously established as core type I interferon signaling components.
Figure 1 shows the workflow and heatmap-style outputs for enriched proximal proteins across the selected pathway members. The authors also used immunoblot validation for selected newly identified proximal proteins, showing that the proximity proteomics workflow produced candidates suitable for deeper biological follow-up.
A key biological finding was the identification of PJA2 as a negative regulator connected to the type I interferon signaling cascade. The study further explored this candidate beyond the discovery dataset, supporting the value of proximity labeling as a starting point for functional mechanism research.
Conclusion
This case shows why proximity labeling proteomics can be valuable for signaling and mechanism research. It can map protein neighborhoods across multiple pathway members, compare stimulated and basal states, and produce candidate regulatory proteins for deeper validation.
For drug discovery and pathway-focused projects, the same logic can support compound-response studies, target deconvolution, and mechanism exploration. The most useful output is not only a protein list, but a controlled and interpretable dataset that helps researchers decide which candidates deserve follow-up experiments.