IP-MS Interactome Profiling Identifies HDAC4/HDAC1/HDAC2 Epigenetic Complex as a DNA Repair Regulator
Di Giorgio E, Dalla E, Tolotto V, D'Este F, Paluvai H, Ranzino L, Brancolini C. (2024) HDAC4 influences the DNA damage response and counteracts senescence by assembling with HDAC1/HDAC2 to control H2BK120 acetylation and homology-directed repair. Nucleic Acids Research, 52(14): 8218–8240.
Study design: Cellular senescence — a state of irreversible cell-cycle arrest — contributes to aging and age-related disease, yet the molecular mechanisms that enforce and maintain senescence remain incompletely understood. The research team at Università degli Studi di Udine investigated the role of HDAC4 in the DNA damage response, hypothesizing that HDAC4 might function in genome maintenance pathways relevant to senescence bypass. They employed Creative Proteomics' immuno-precipitation proteomics (IP-MS) service to characterize the HDAC4 interactome, identifying HDAC1 and HDAC2 as the core components of a novel epigenetic complex that regulates DNA double-strand break repair pathway choice.
Key results: The IP-MS analysis revealed that HDAC4 physically interacts with HDAC1 and HDAC2 to form a deacetylase complex that removes acetyl groups from histone H2B at lysine 120 (H2BK120ac). This HDAC4/HDAC1/HDAC2 complex modulates homology-directed repair (HDR) efficiency through dynamic deacetylation of H2BK120. When HDAC4 is degraded during Ras-induced senescence, this complex disassembles, H2BK120ac accumulates, BRCA1 and CtIP recruitment to DNA damage sites is impaired, and cells accumulate unrepaired DNA damage — which in turn reinforces the senescence transcriptional program. Forced HDAC4 expression reduces γH2AX genomic spreading and partially restores DNA repair capacity. The study establishes HDAC4 as a potential therapeutic target for modulating senescence and DNA repair in cancer and aging, and demonstrates that the HDAC4/HDAC1/HDAC2 complex represents a node for pharmacological intervention in senescence-associated pathologies.
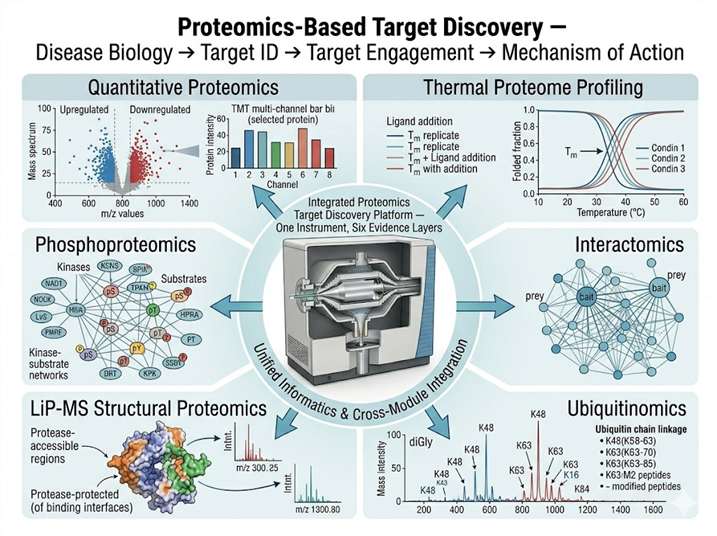
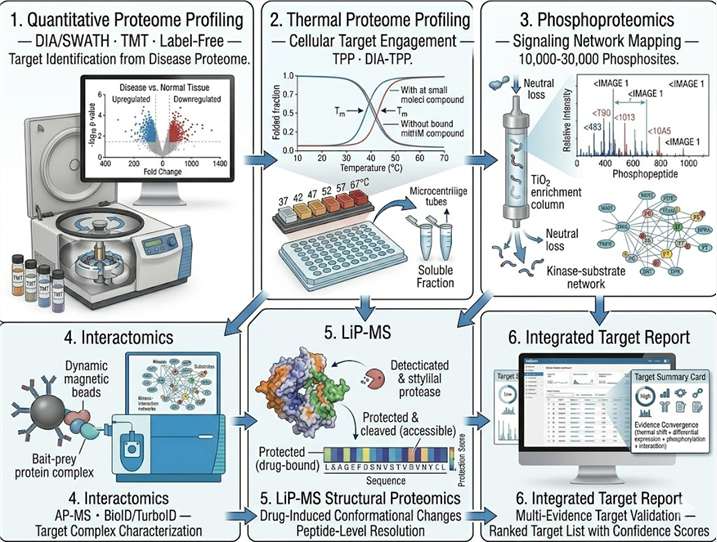
Relevance to our proteomics-based target discovery platform: This study exemplifies the integrated target discovery logic that our platform operationalizes: interactomics (IP-MS identification of HDAC4's binding partners, revealing the HDAC1/HDAC2 complex as the functional unit), mechanistic proteomics (linking interactome data to a specific post-translational modification — H2BK120ac — and a specific biological process — HDR), and target validation (demonstrating that loss of the target complex produces a disease-relevant phenotype — senescence — and that restoration of the target partially rescues normal function). For target discovery programs seeking to move beyond differential expression lists to mechanistically validated targets, our integrated proteomics platform provides the same workflow: AP-MS or proximity-labeling MS to define the target's interactome, phosphoproteomics or modification-specific proteomics to map the signaling context, and thermal profiling or LiP-MS to confirm that the target can be engaged by small molecules — all on a single, harmonized MS platform.