Interactome and Network Target Identification Service
Five integrated service lines for comprehensive protein interaction network mapping and druggable target discovery — from AP-MS and BioID through XL-MS, HDX-MS, and LiP-MS.
Your team has a protein of interest — a disease-associated kinase from a GWAS study, a receptor implicated in tumor progression, or a hit from a phenotypic screen. The more important question for drug discovery is: which of its interaction partners represent druggable intervention points in the disease network?
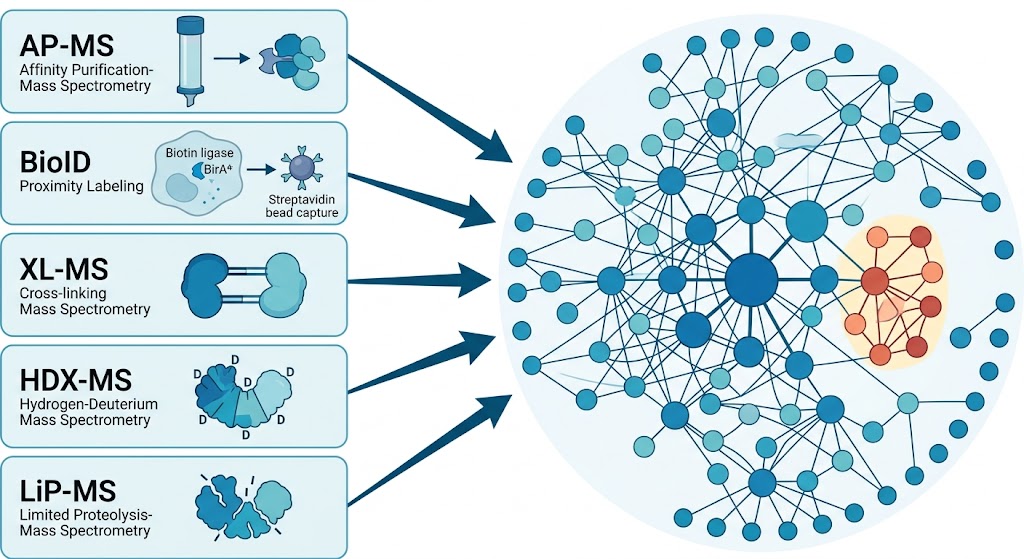
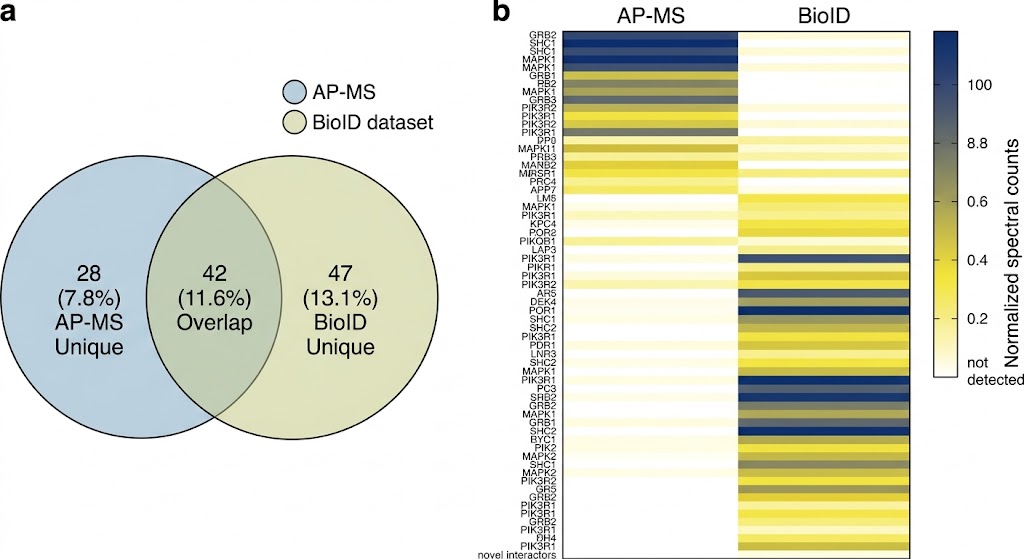
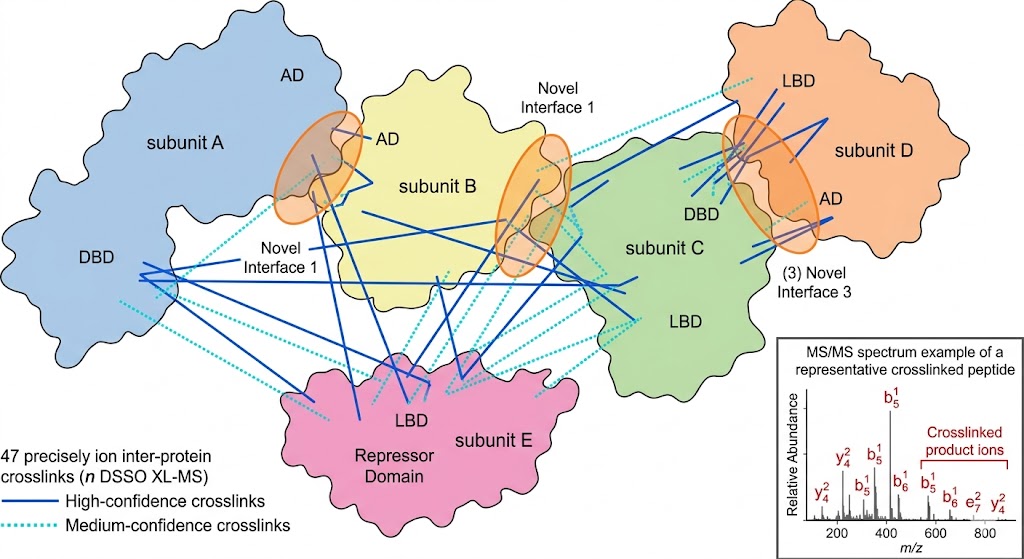
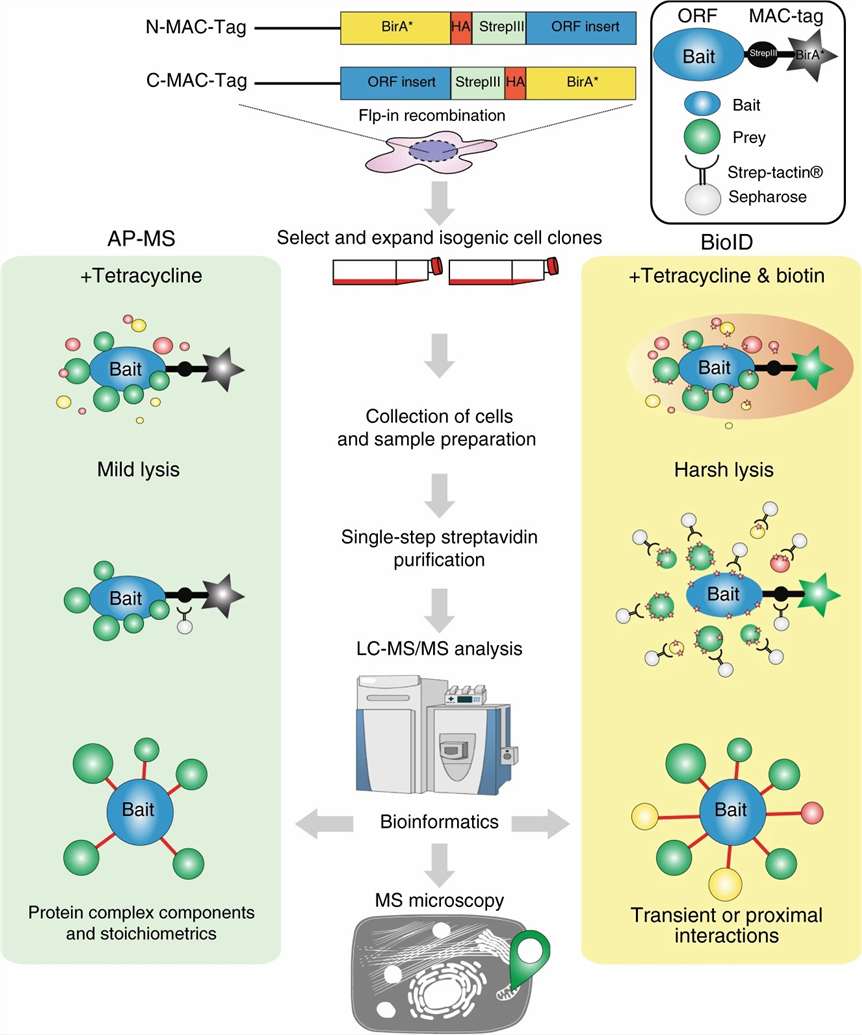
The MassTarget platform addresses this challenge through five complementary interactome service lines — Affinity Purification MS (AP-MS), Proximity-Dependent Biotinylation (BioID/TurboID), Cross-Linking MS (XL-MS), HDX-MS for Epitope Mapping, and Limited Proteolysis MS (LiP-MS) — each designed to answer a specific type of protein interaction question.
Key Advantages:
- Five integrated interactome methods — AP-MS, BioID, XL-MS, HDX-MS, and LiP-MS
- Dedicated BioID/TurboID pipeline for membrane proteins and transient interactors
- XL-MS and HDX-MS for structural interaction details
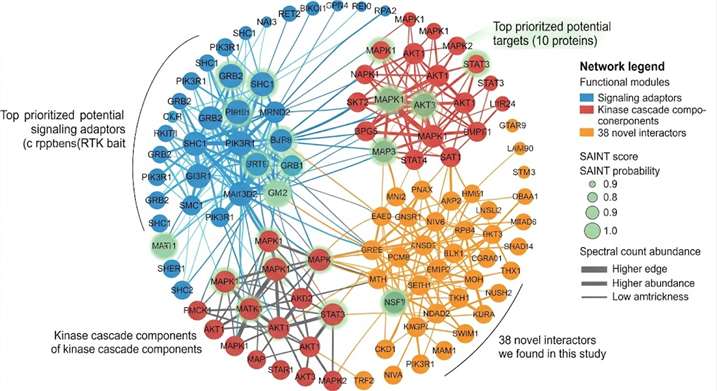
- Full bioinformatics with SAINT scoring and network visualization
- Biophysical validation (BLI/SPR) available as integrated downstream step