Protein Microarray with MS Readout for Label-Free Target Identification
Proteome-wide target fishing without compound modification — combining protein microarray coverage with mass spectrometry identification power.
Finding the protein target of a bioactive compound is often the hardest part of early drug discovery. Traditional approaches such as pull-down proteomics and chemical proteomics require labeled probes, antibody development, or functional assays — each adding time, cost, and potential artifacts.
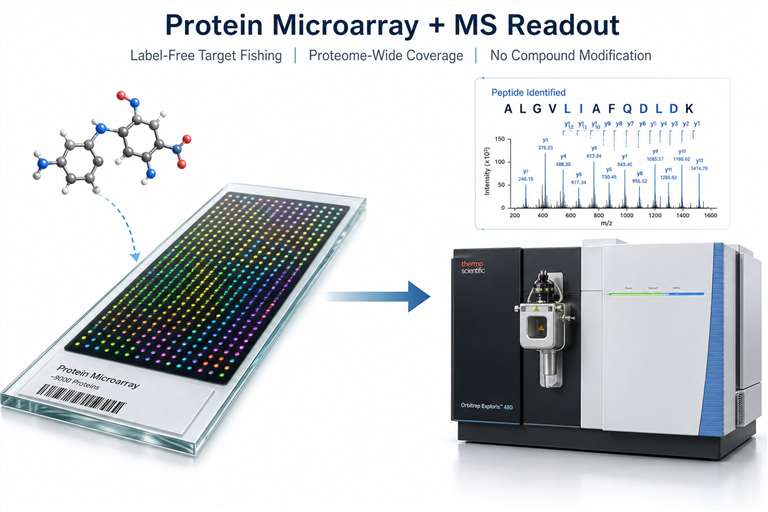
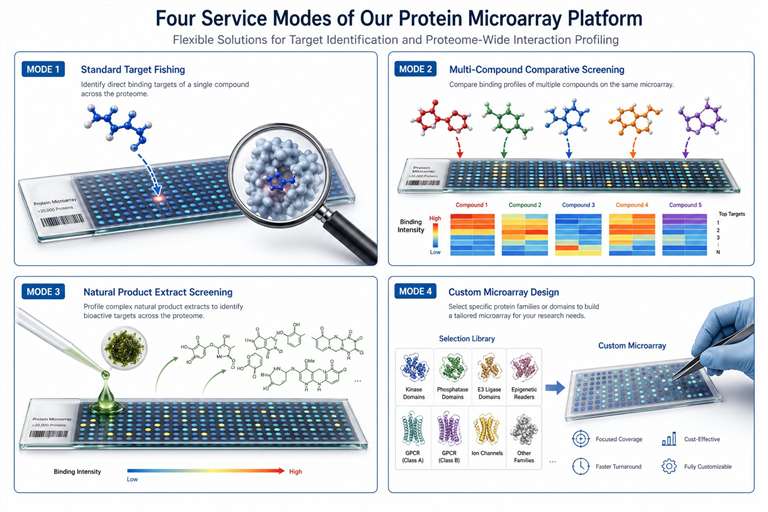
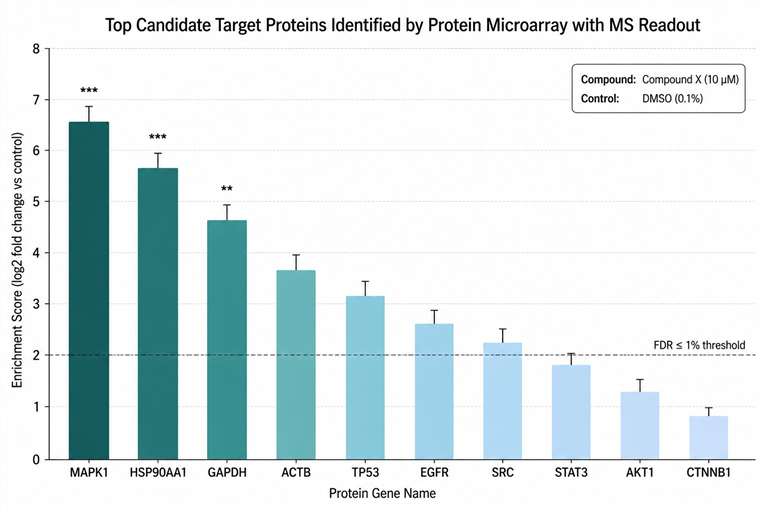
At Creative Proteomics, our Protein Microarray with MS Readout service gives you a direct, label-free path from compound to target. We immobilize thousands of individually purified proteins on a single microarray slide, incubate your compound directly, and identify bound proteins by LC-MS/MS — delivering a ranked list of candidate targets with peptide-level evidence.
Key Advantages:
- No compound modification required — use your molecule as-is, including natural product extracts and crude mixtures.
- Proteome-wide coverage — 10,000–20,000+ purified proteins screened in a single experiment.
- MS-based identification — every hit backed by sequenced peptides with FDR-controlled confidence.
- Works with challenging compounds — covalent binders, fragments, peptides, and unpurified samples.