Overcoming the Bottlenecks of Covalent FBLD
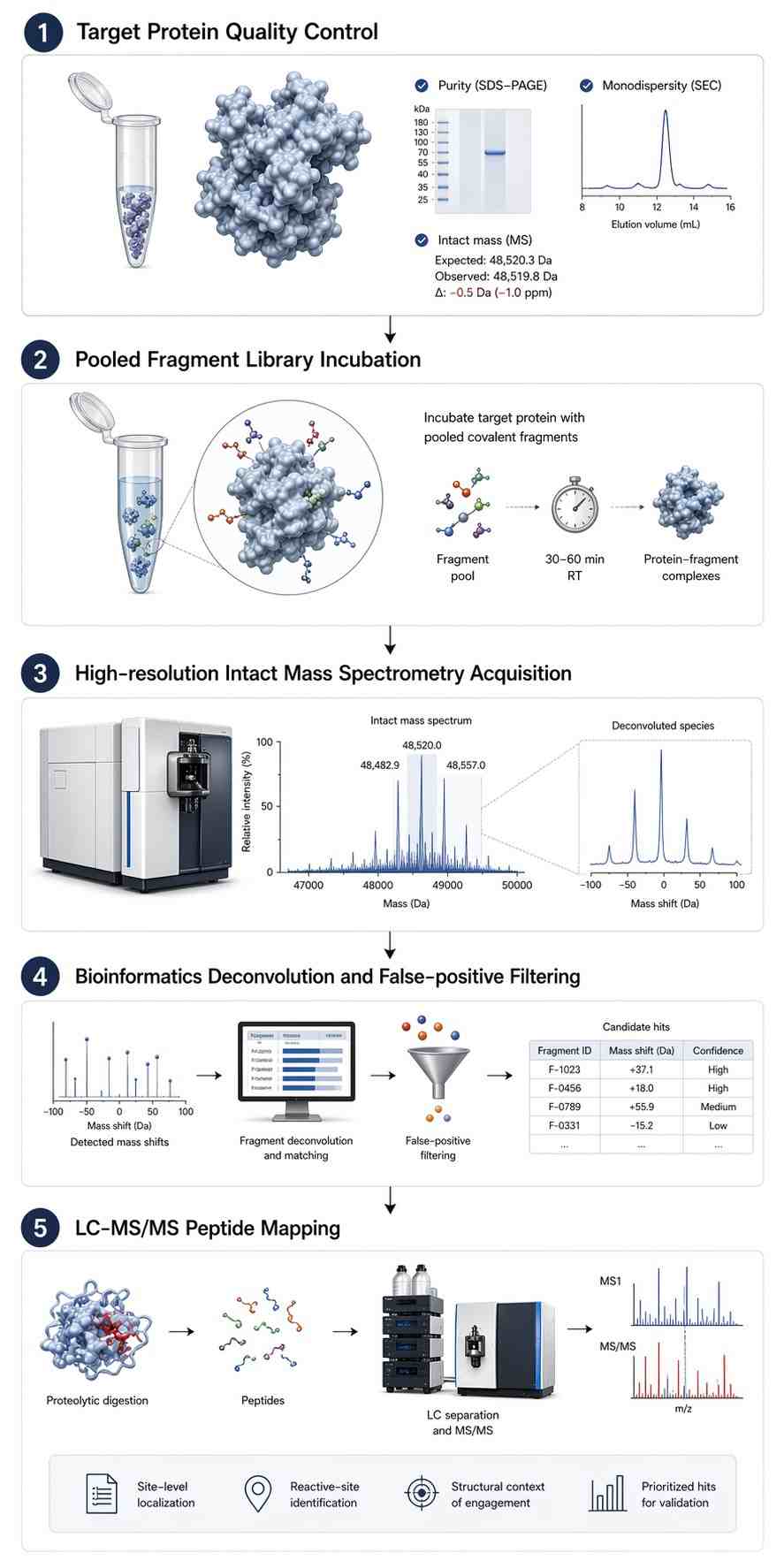
Fragment-based drug discovery (FBLD) has become a cornerstone of modern pharmaceutical research, particularly when confronting targets that lack deep, well-defined binding pockets. By exploring chemical space with low-molecular-weight molecules (typically<300 Da), researchers can identify highly efficient starting points for drug development. However, developing covalent fragments—designed to form an irreversible or reversible permanent bond with a target amino acid residue—presents immense analytical challenges that traditional screening methods struggle to resolve.
Standard biophysical techniques, such as Thermal Shift Assays (TSA) or Surface Plasmon Resonance (SPR), provide indirect evidence of binding. While historically useful, these methods are notorious for generating high false-positive rates when dealing with covalent binders. At the high concentrations required for fragment screening (often in the millimolar range), compounds may cause a thermal shift simply by non-specifically aggregating on the protein surface, rather than forming a true, functional covalent bond. Conversely, specific but structurally subtle covalent binders might be missed entirely (false negatives) if their binding does not induce a substantial change in the target protein's global thermodynamic stability.
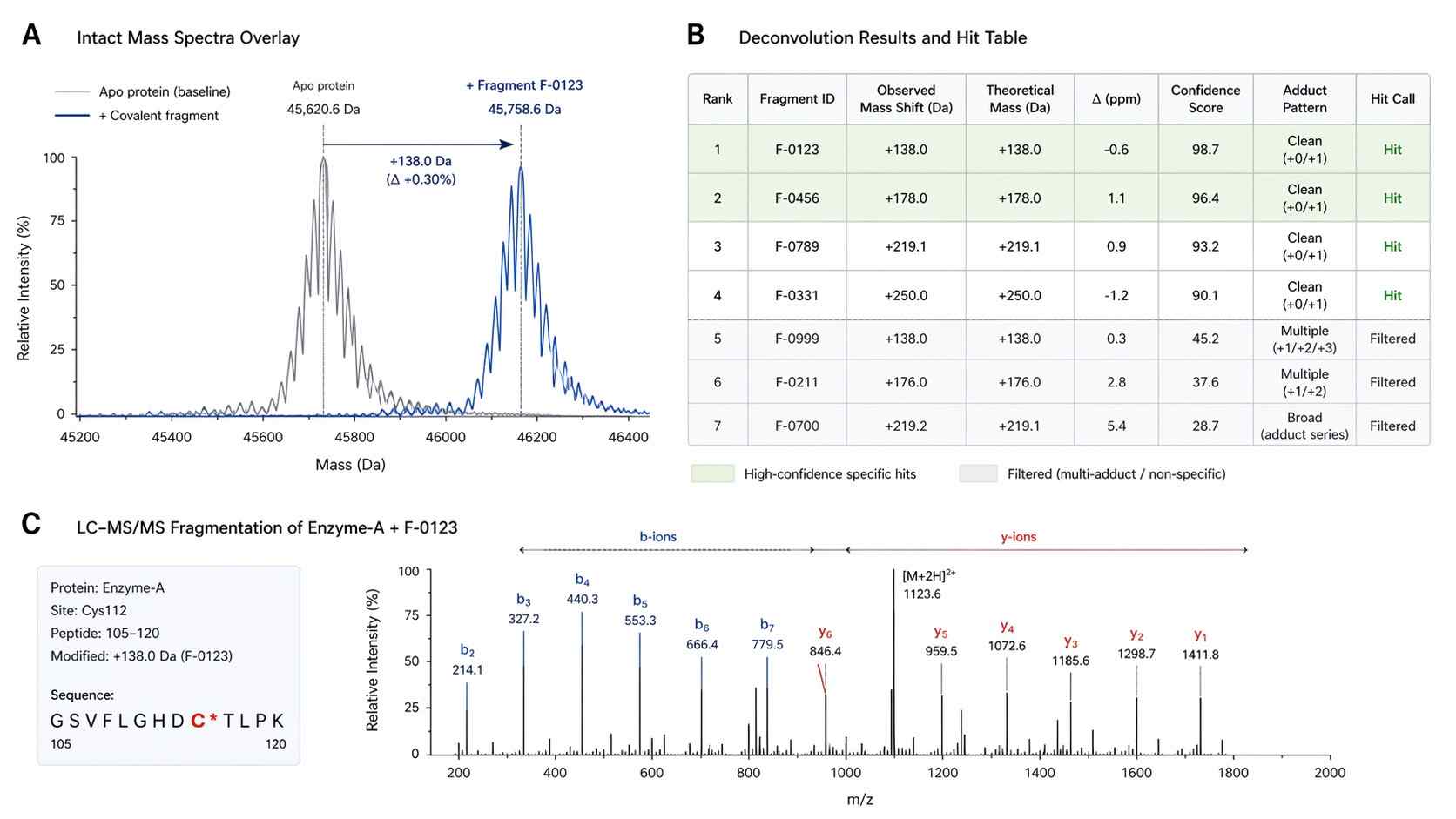
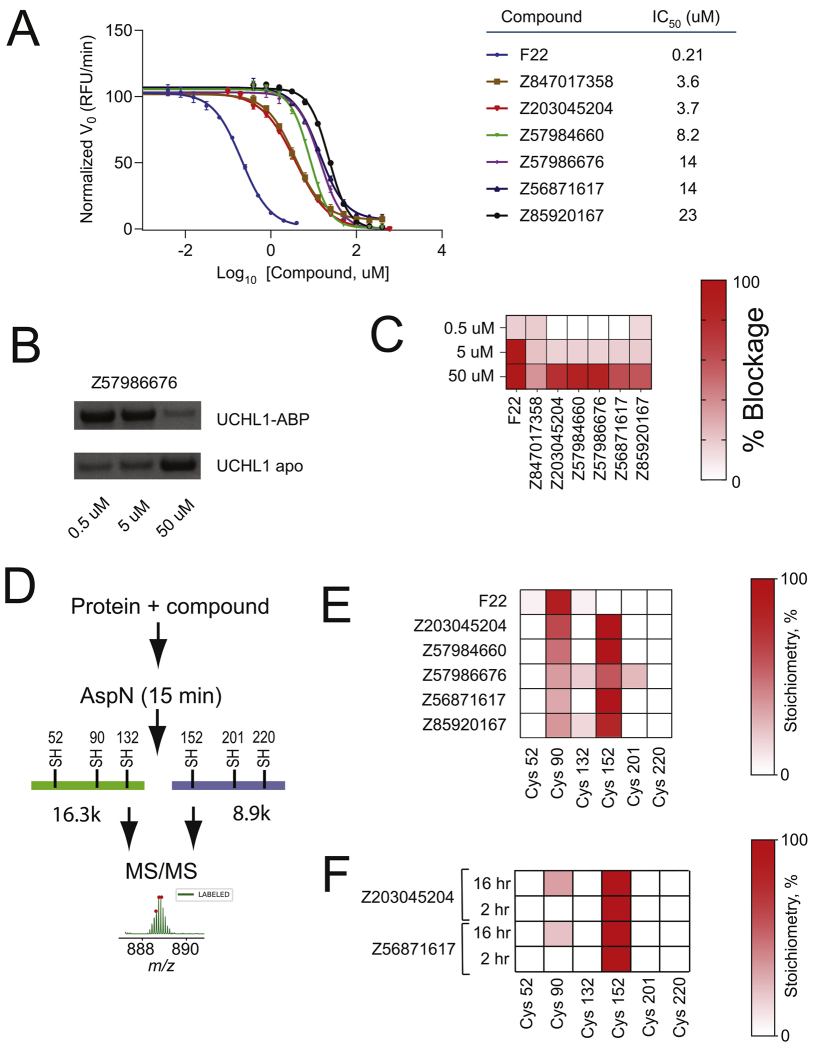
Mass spectrometry (MS) revolutionizes this process by offering a direct, label-free readout of the binding event. By measuring the exact monoisotopic mass of the target protein before and after incubation with a fragment library, we can observe the precise "mass shift" (+ΔMass) that occurs exclusively when a covalent bond is formed. This approach eliminates the reliance on reporter dyes, complex assay development, or indirect stability measurements, providing a definitive, evidence-based path forward for medicinal chemists.