Drug Discovery Solutions — From Disease Biology to Validated Leads
A single mass spectrometry partner across your entire discovery pipeline — no handoffs, no fragmented data, no assay transfer delays.
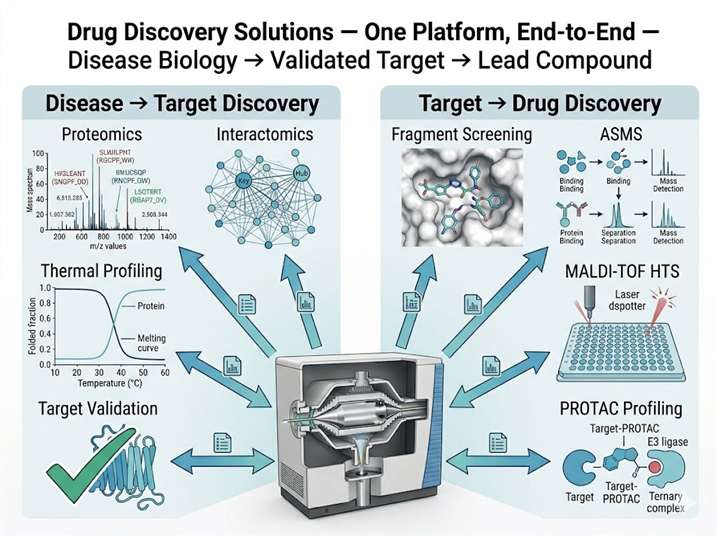
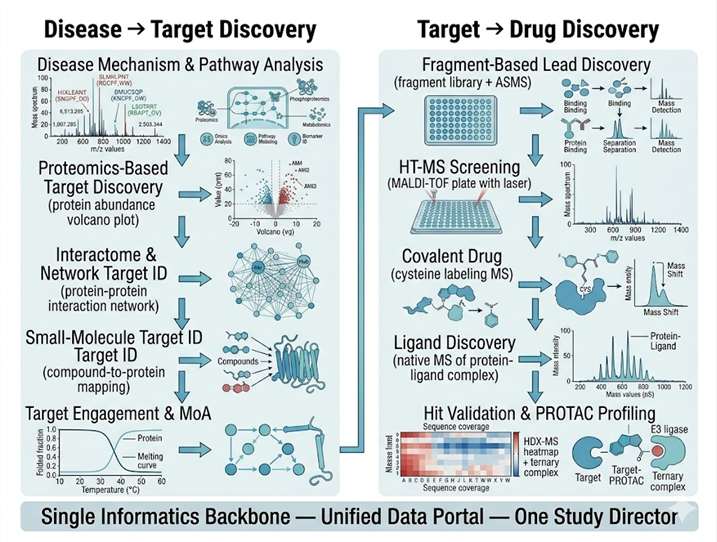
Drug discovery demands integration: the target identified by proteomics must connect seamlessly to the binding assay that confirms ligand engagement; the hit from a high-throughput screen must be validated by the same MS platform that will characterize its mechanism. Yet most discovery programs piece together data from separate CROs — one for proteomics, another for screening, a third for structural biology — losing weeks to assay transfer and months to reconciling incompatible data formats.
Our Drug Discovery Solutions platform closes this gap. We operate the full spectrum of mass spectrometry technologies under one roof — from discovery proteomics and interactomics for target identification through affinity selection MS, MALDI-TOF HTS, and fragment-based screening for hit finding, to covalent binding characterization and PROTAC complex profiling for lead optimization. Every dataset shares a common informatics backbone, every result transfers directly to the next stage, and every decision is supported by the same analytical team that generated the underlying data. The result is a discovery pipeline where the transition from target to hit to lead is measured in weeks, not quarters.