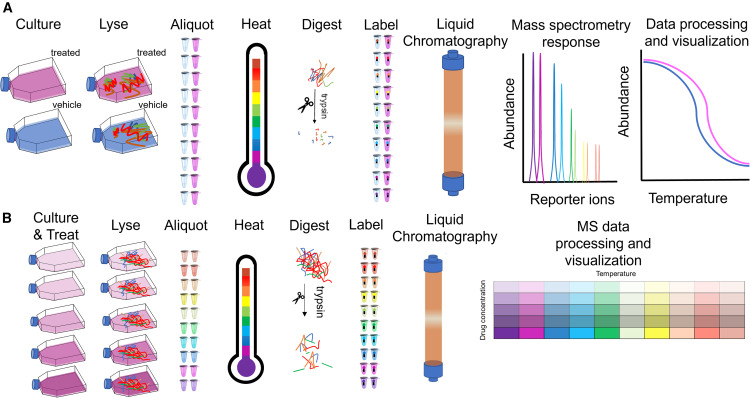
Our Dual-Engine Platform: TPP & LiP-MS Orthogonal Validation
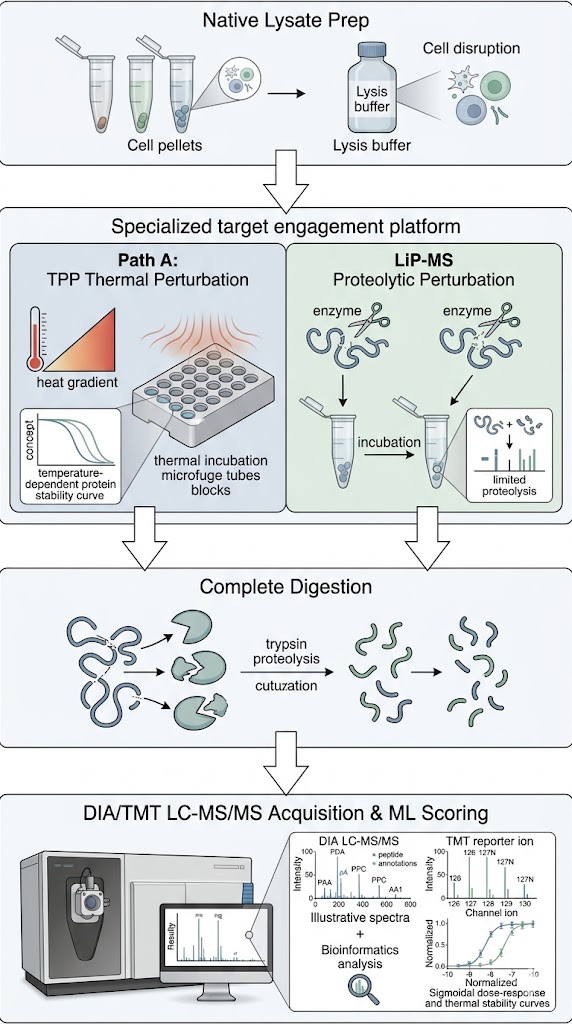
Not all proteins behave the same way when a drug binds to them. If you rely on just one type of stability assay, you risk missing your target entirely. To provide you with the highest possible confidence and minimize the risk of false negatives, we have built a "Dual-Engine" orthogonal screening platform. We interrogate your samples using two fundamentally different physical principles.
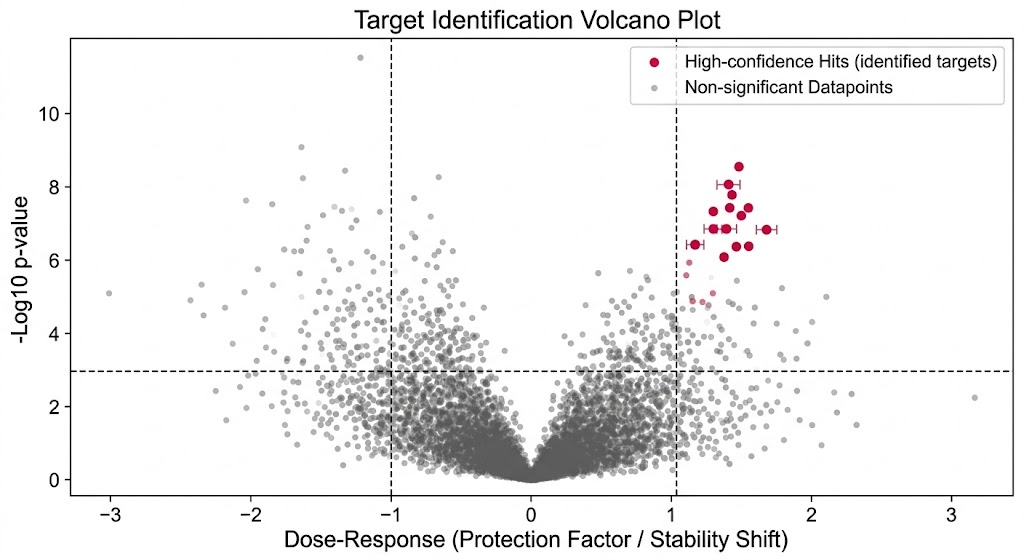
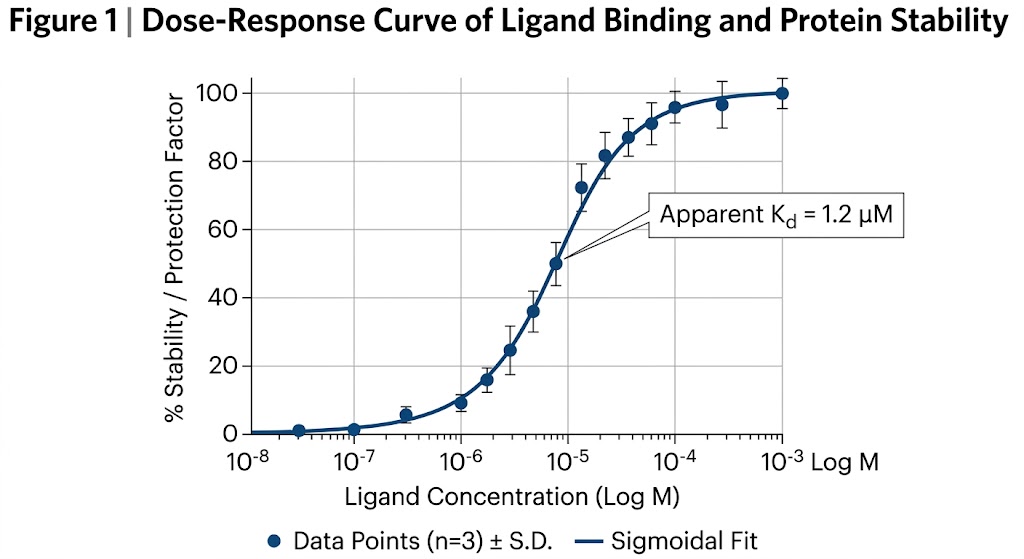
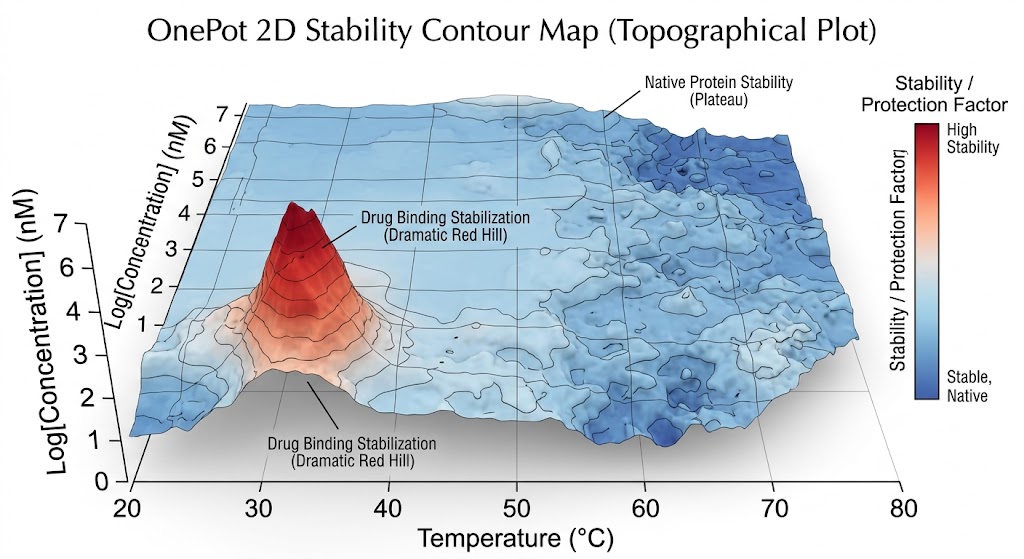
First, we utilize Thermal Proteome Profiling (TPP). Also known as MS-based Proteome-wide Thermal Stability Profiling coupled to MS, this approach applies a gradient of heat to the cell lysate. Proteins naturally unfold and precipitate as the temperature rises. When your drug binds to its target, it acts like a structural anchor, significantly shifting the temperature at which that specific protein melts. We use multiplexed mass spectrometry to track the melting curves of thousands of proteins simultaneously, flagging the ones that stabilize in the presence of your drug.
Second, we utilize Limited Proteolysis–MS (LiP-MS). Some highly flexible proteins, or proteins binding allosteric modulators, may not show a noticeable shift in their overall melting temperature. For these challenging targets, LiP-MS is the perfect solution. Instead of heat, we use a broad-specificity protease to gently probe the protein's surface. When your drug binds, it physically covers the binding pocket or induces a conformational change that hides specific digestion sites. By measuring these precise proteolytic protection patterns, we can identify targets that thermal methods miss, while also providing valuable clues about the exact location of the binding site.