Feature-Based Molecular Networking (FBMN) Service
Quantitative, multi-tool compatible molecular networking for untargeted metabolomics.
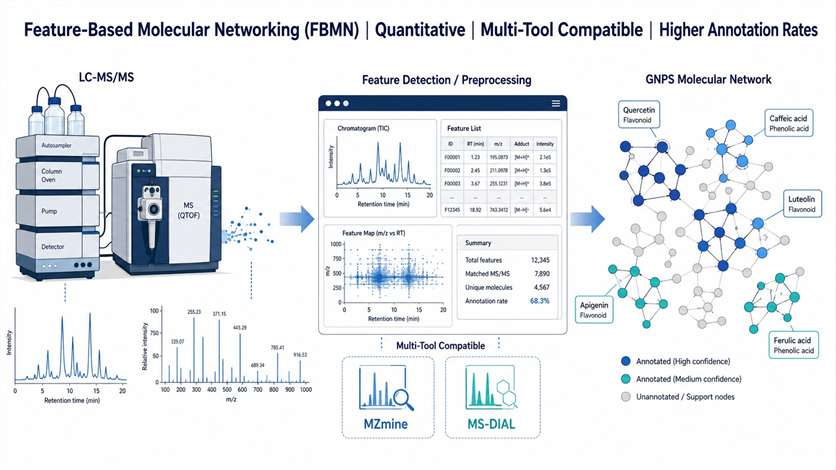
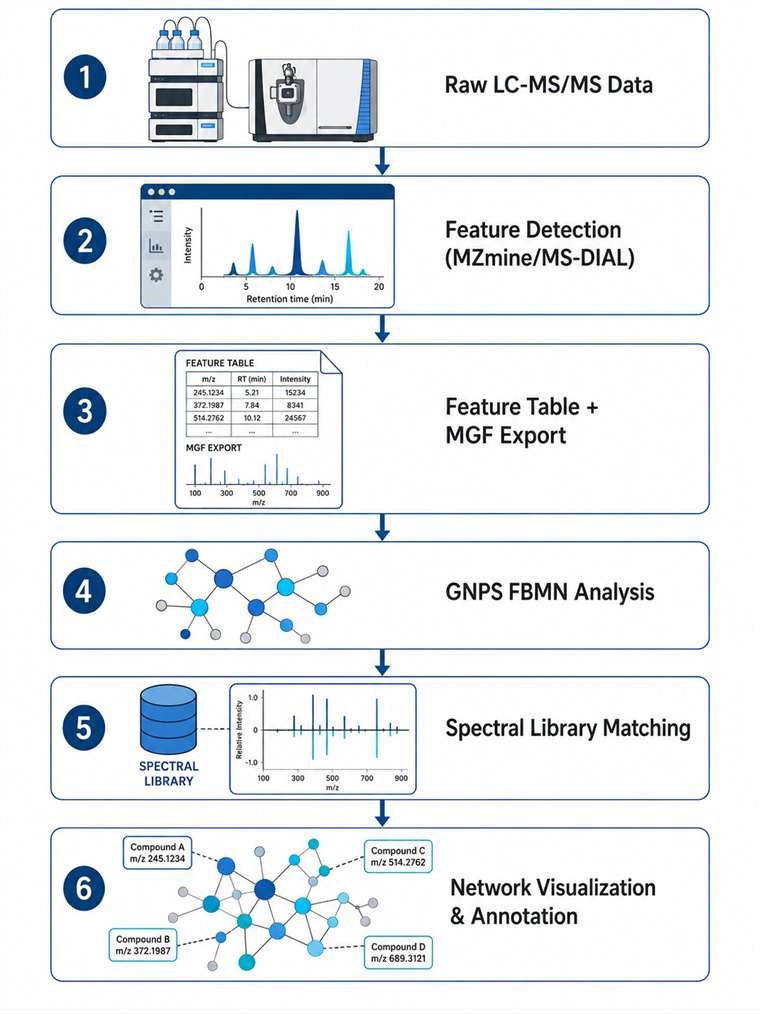
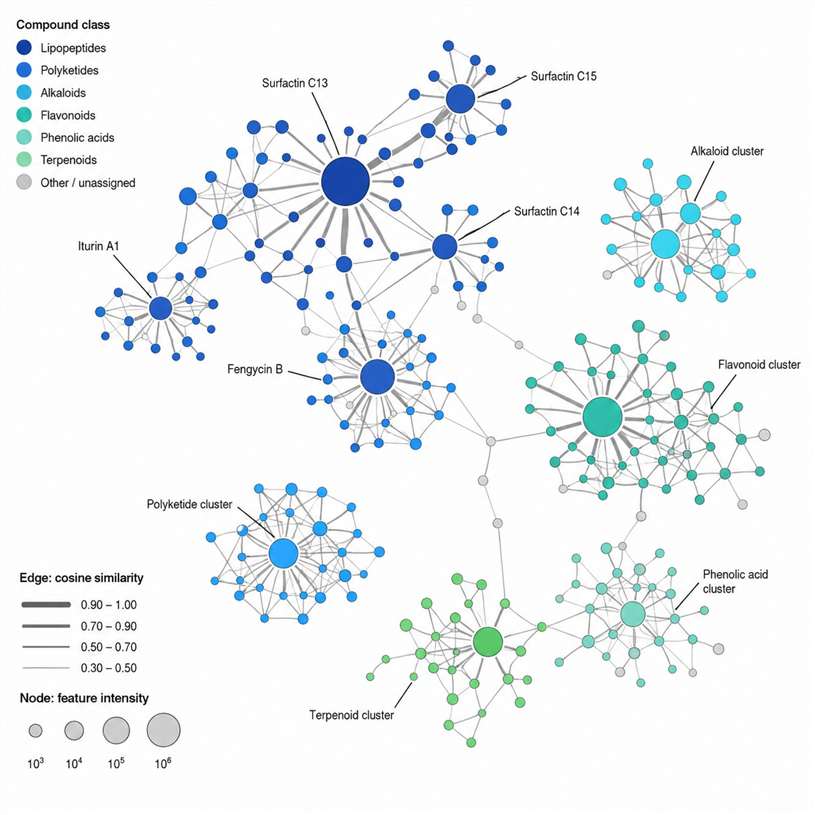
Feature-Based Molecular Networking (FBMN) is an advanced computational workflow that bridges popular LC-MS/MS data processing tools with the GNPS molecular networking environment. Unlike classical molecular networking, FBMN retains quantitative feature information — retention time, accurate mass, and intensity — from the preprocessing stage, enabling more accurate network construction, isomer resolution, and direct integration with statistical analysis.
At Creative Proteomics, we provide end-to-end FBMN analysis services for researchers working with untargeted metabolomics, natural product discovery, and MS-based chemical profiling. Our team handles the entire pipeline — from raw data preprocessing through feature detection, GNPS networking, spectral library matching, and biological interpretation — so you can focus on the science.
Key Advantages:
- Quantitative feature retention for accurate network-based comparisons across sample groups.
- Compatibility with MZmine 2/3, MS-DIAL, XCMS, Progenesis QI, MetaboScape, and OpenMS.
- Support for isomer resolution, ion mobility spectrometry (IMS) data, and MSE acquisition.
- Integrated statistical analysis with group-level comparisons and visualization.