Integrated LiP-MS + TPP for Orthogonal Drug Target Deconvolution
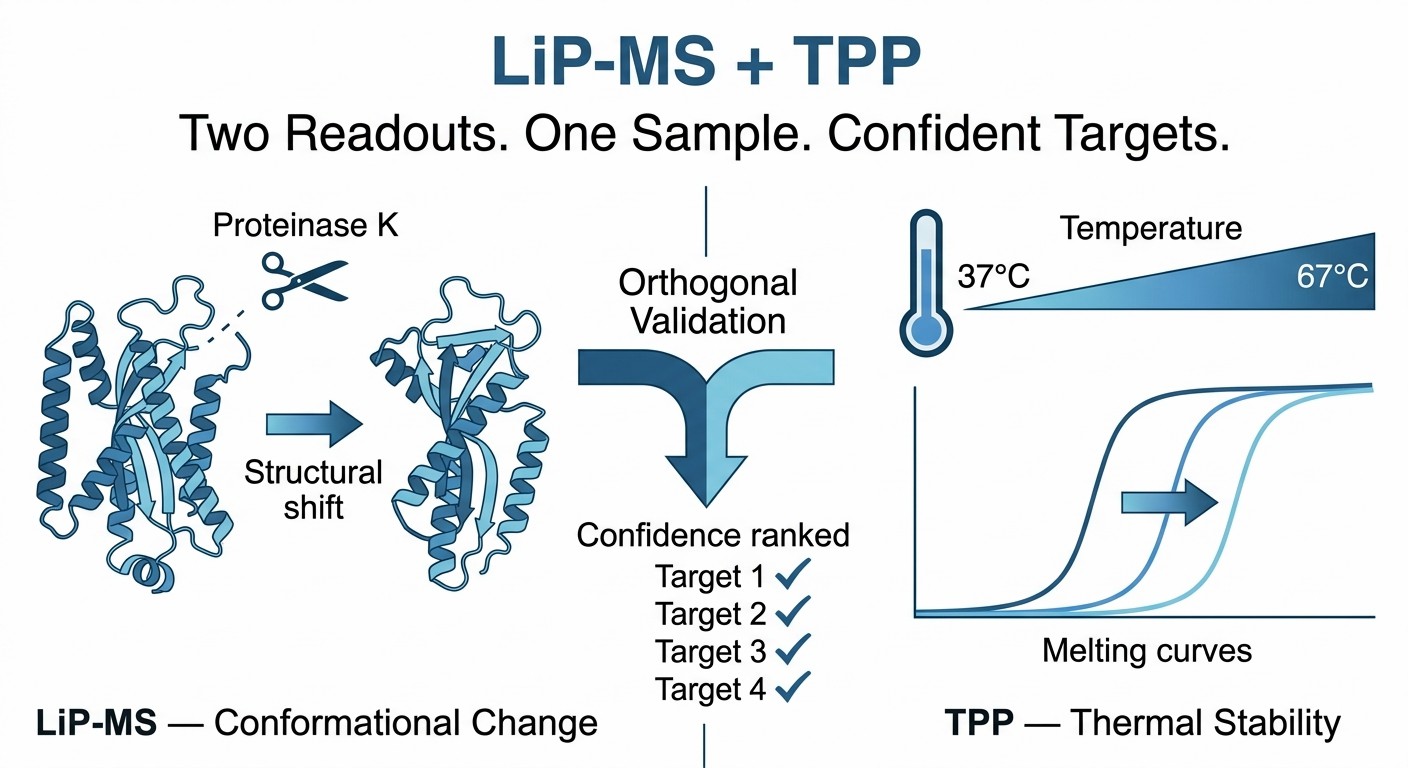
Two orthogonal chemoproteomic readouts — conformational dynamics and thermal stability — from a single biological sample.
Every label-free chemoproteomic method has blind spots. TPP detects drug-induced thermal stability shifts but cannot report conformational dynamics. LiP-MS detects changes in protease accessibility but may miss targets where binding does not alter local structure. Relying on either method alone risks false negatives.
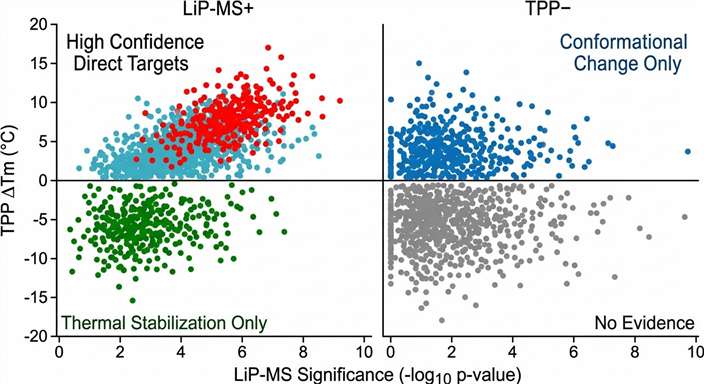
The solution is not to choose one method over the other — it is to run them together. LiP-MS and TPP are orthogonal by design. LiP-MS reads out structural dynamics at peptide-level resolution; TPP reads out global stability changes at protein-level resolution. When a candidate target is flagged by both methods, the confidence in direct engagement rises dramatically. When flagged by only one, the result still provides valuable mechanistic information — a conformational change without stability shift, or a thermal stabilization without detectable structural rearrangement.
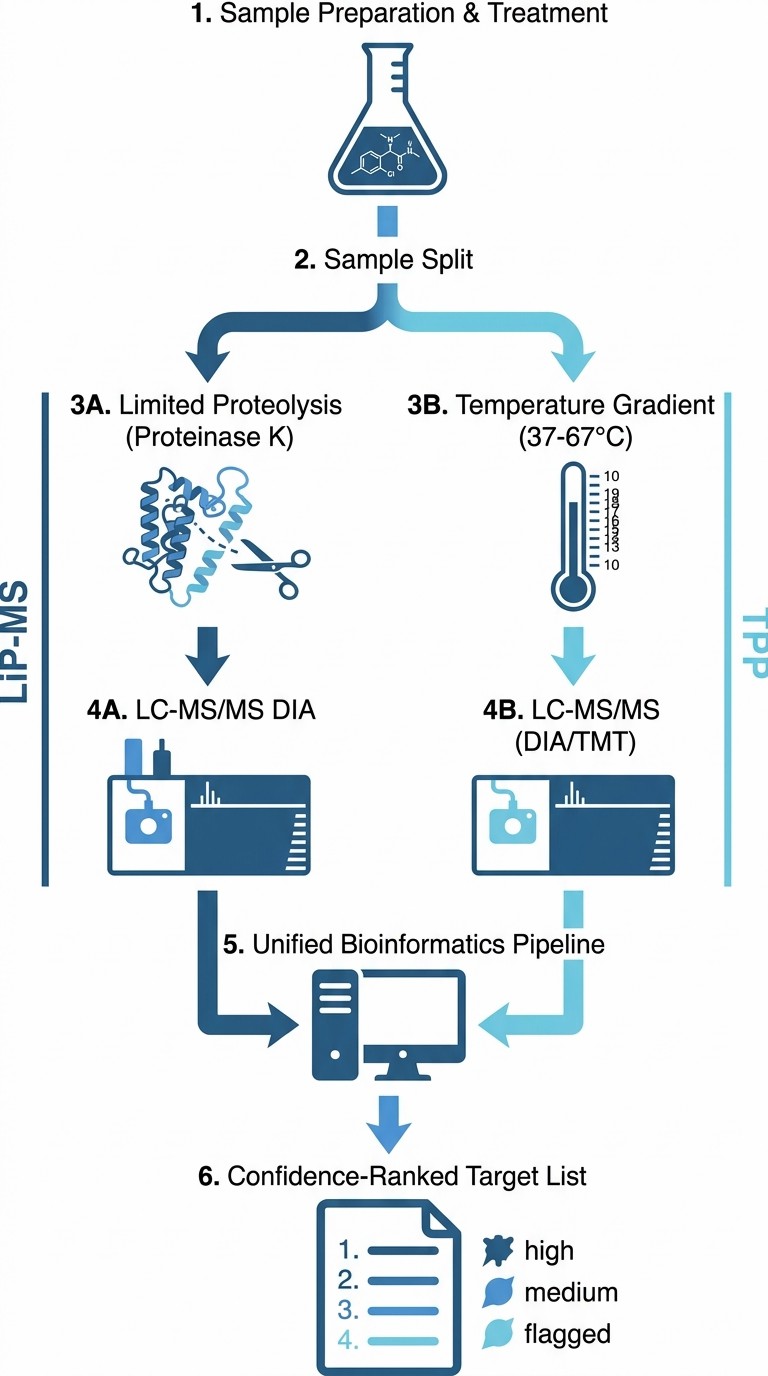
We offer LiP-MS and TPP as an integrated, coordinated workflow — not two separate assays run independently. From a single compound-treated sample, we generate two independent lines of evidence, cross-correlate them through a unified bioinformatics pipeline, and deliver a confidence-ranked target list. This approach has been validated in published studies: a 2024 study in Nature Chemical Biology showed >60% concordance between LiP-MS and TPP across the E. coli and human proteomes, demonstrating that the two methods provide complementary target identification data.
Our service is part of the broader multi-omics integration platform at Creative Proteomics, and complements our standalone LiP-MS service and standalone TPP service by adding the orthogonal validation that single-method approaches cannot provide.