Fragment-Based Lead Discovery by Native MS — Label-Free Fragment Screening and Hit Validation
From fragment to lead: native mass spectrometry reads the protein-fragment complex directly, without labels, immobilisation, or chromogenic substrates.
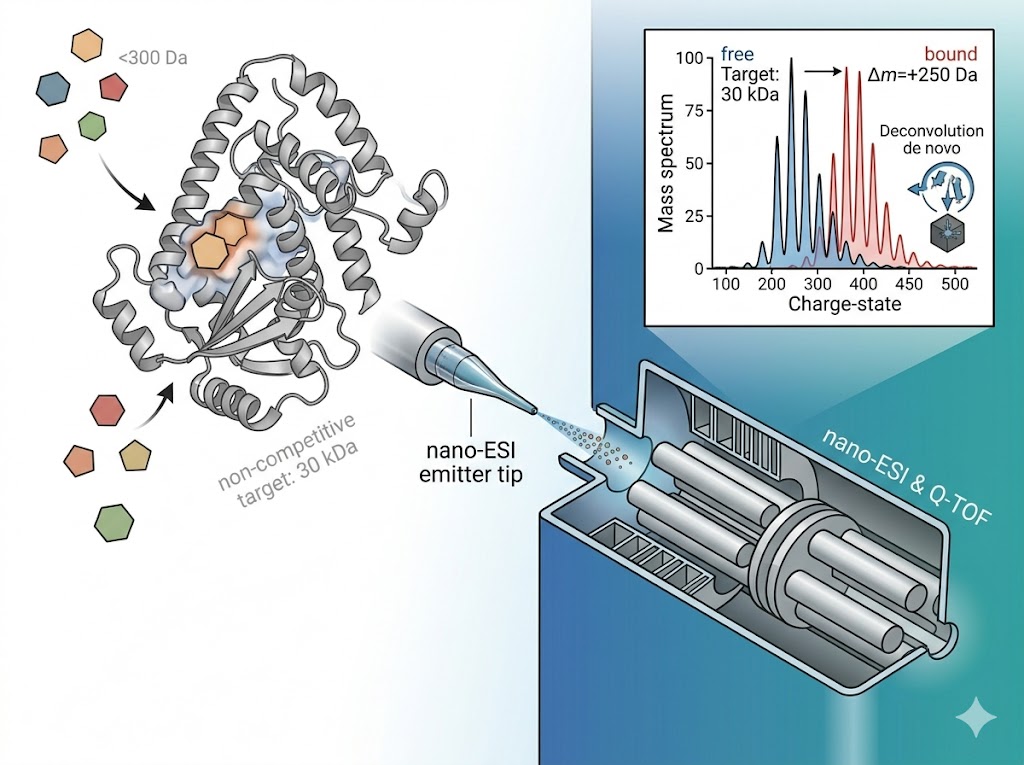
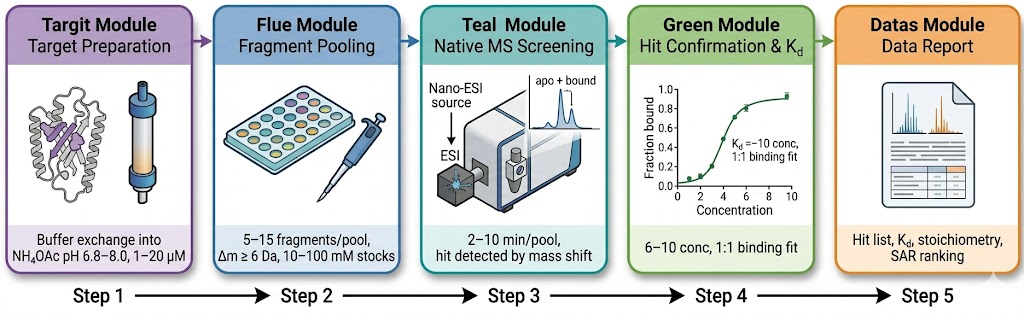
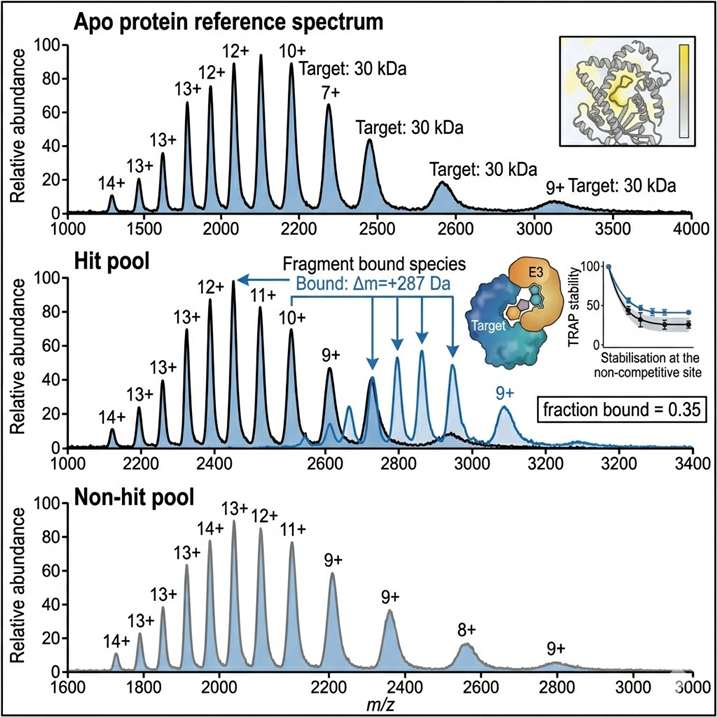
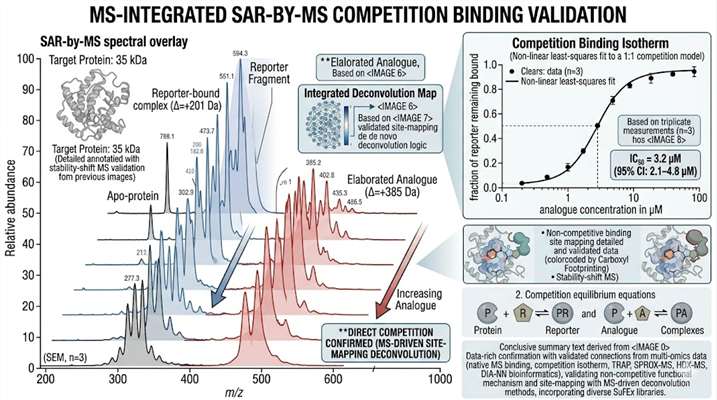
Fragment-based lead discovery (FBLD) identifies low-molecular-weight (typically <300 Da) compounds that bind to a target protein with weak but efficient binding, then elaborates them into potent leads through iterative medicinal chemistry. The central analytical challenge at the heart of every FBLD campaign is detection — fragment binding is typically weak (Kd from high µM to low mM), and the small molecular weight of fragments provides minimal spectroscopic signal for conventional biochemical assays. Native mass spectrometry addresses this challenge by detecting the intact protein-fragment complex directly: the mass of the complex is measured precisely enough to confirm binding, and the fraction of protein in the bound state is quantified to determine binding affinity. Our FBLD service by native MS covers the full fragment-to-lead trajectory — primary fragment library screening, hit validation by dose-response Kd determination, SAR-by-MS for fragment elaboration, and covalent fragment profiling.
Key Advantages:
- No assay development required — the masses of the target and fragments are the only information needed to start screening.
- Direct binding stoichiometry from every measurement — distinguishes specific 1:1 engagement from higher-order or non-specific aggregation.
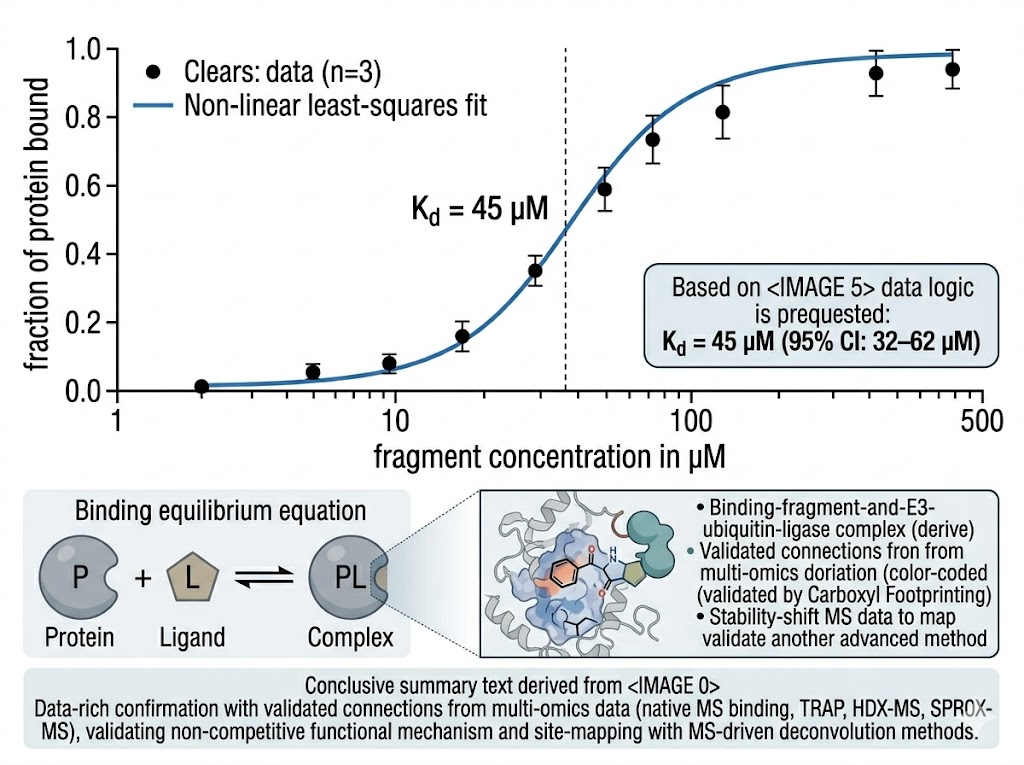
- Kd determination by native MS titration across nM to mM affinity range, without surface immobilisation artefacts.
- SAR-by-MS competition binding for rapid analogue ranking during fragment elaboration without assay redevelopment.
- Compatible with challenging targets — membrane proteins, protein-protein interaction interfaces, intrinsically disordered proteins, and multi-protein complexes.
- Low protein consumption — 50–200 µg per 1,000–5,000 fragment set, comparable to or less than SPR and substantially less than NMR.