Xu R, Yu H, Wang Y, et al. "Natural product virtual-interact-phenotypic target characterization: A novel approach demonstrated with Salvia miltiorrhiza extract." Journal of Pharmaceutical Analysis, 2024. DOI: 10.1016/j.jpha.2024.101101
Background
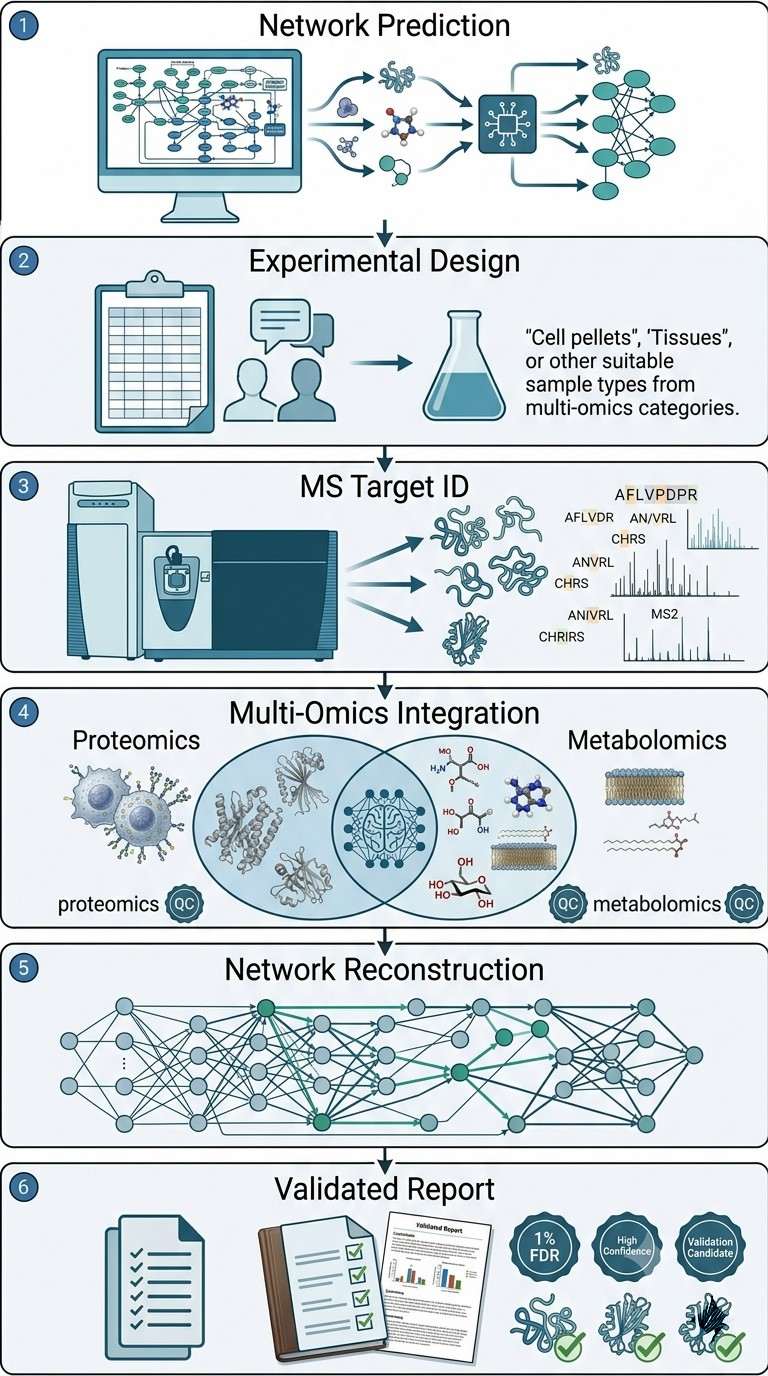
Natural product target identification has two hard problems: extracts contain hundreds of constituents, and there is no standard workflow to connect computational predictions to experimental proof. Xu et al. (2024) addressed this with the Natural Product Virtual-Interact-Phenotypic (NP-VIP) strategy, which integrates virtual screening, chemical proteomics, and metabolomics to characterize targets of Salvia miltiorrhiza extract — a botanical drug used for ischemic stroke.
Methods
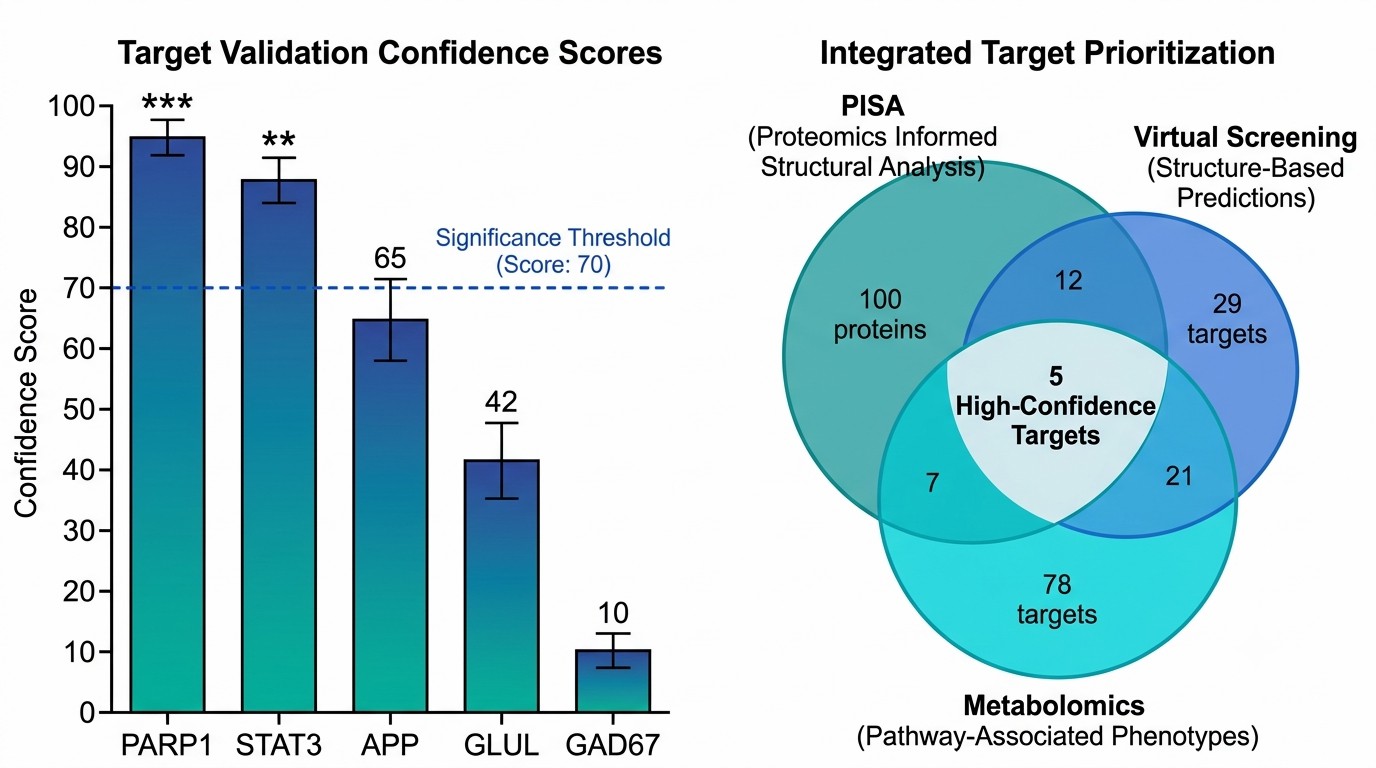
The team used a three-pronged design. First, 75% ethanol extraction followed by silica gel column chromatography and MS-based profiling identified 151 compounds in the extract. Second, computational target prediction using structural and bioactivity similarity tools, cross-referenced against DisGeNET, produced 29 high-confidence predicted targets. Third, two orthogonal MS approaches ran in parallel: PISA (thermal stabilization assay) for unbiased proteome-wide binding detection (100 differential proteins) and untargeted metabolomics for dose-dependent metabolite profiling (82 metabolites linked to 78 metabolic enzyme targets).
Results
Cross-comparison of the three datasets converged on five high-confidence targets: PARP1, STAT3, APP, GLUL, and GAD67. These span cell death signaling (PARP1), inflammatory response (STAT3), amyloid processing (APP), and glutamate metabolism (GLUL, GAD67) — all relevant to ischemic stroke pathology. DARTS combined with Western blot independently confirmed that the extract directly binds all five proteins.
Conclusion
The NP-VIP strategy showed that integrating computational network pharmacology with orthogonal MS approaches can systematically resolve the multi-target mechanism of a complex natural product extract. The same workflow applies broadly to natural product and multi-target drug discovery projects.