Molecular Glue Target Identification Service — MS-Based Neosubstrate Discovery from Phenotypic Hits to Preclinical Leads
Your molecular glue was designed to bring an E3 ligase and an unknown target together. The critical question — what is the target?
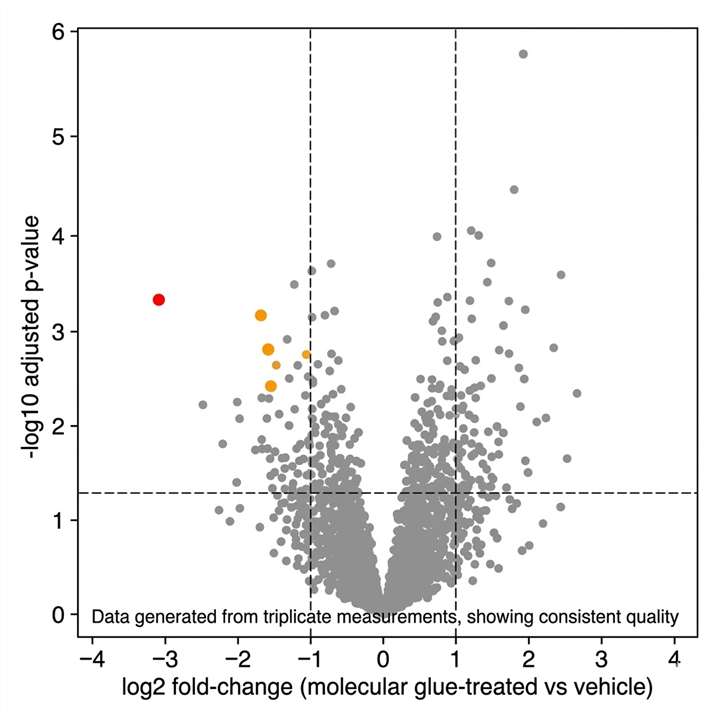
Molecular glues are monovalent small molecules that induce or stabilise protein-protein interactions between an E3 ubiquitin ligase and a previously non-interacting neosubstrate protein, leading to ubiquitination and proteasomal degradation of the neosubstrate. Unlike PROTACs — where both the target and E3 ligand are explicitly designed into a bifunctional molecule — molecular glues work by binding to the E3 ligase and modifying its surface to create a neo-interface that recruits a previously unrelated protein. This means that for most molecular glue programmes, the identity of the degraded target is unknown at the outset and must be discovered empirically.
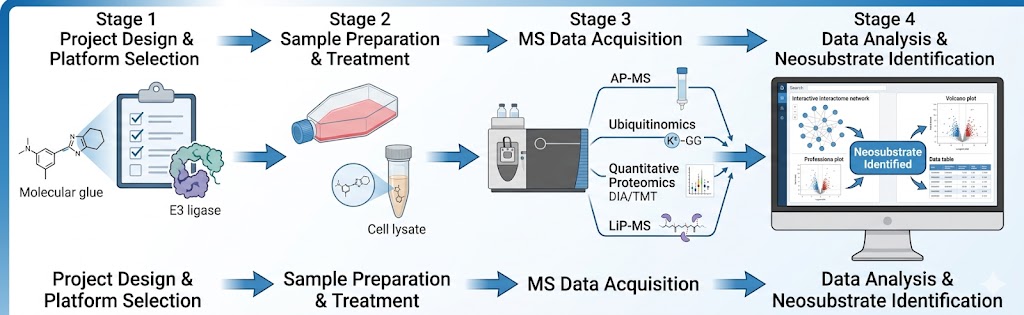
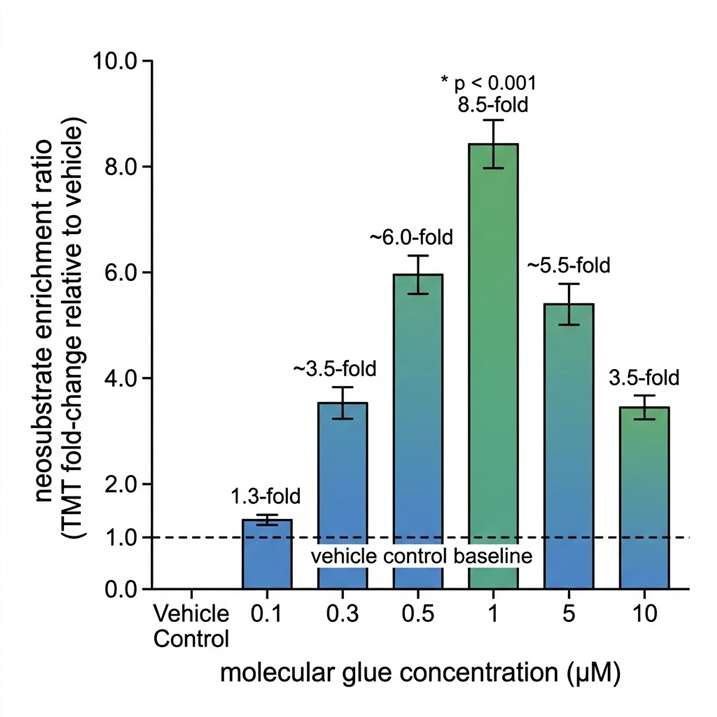
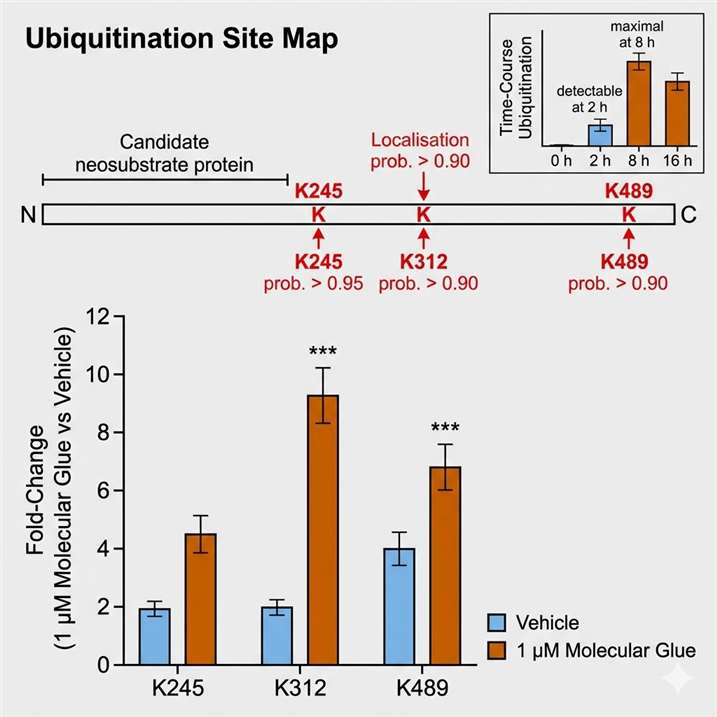
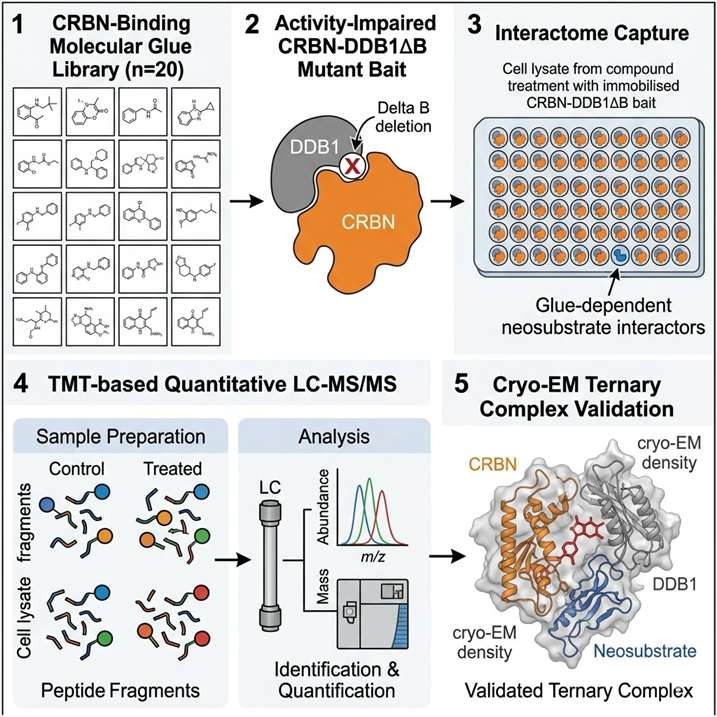
Mass spectrometry-based proteomics is the only methodology capable of identifying the neosubstrate of a molecular glue in an unbiased, proteome-wide manner — without requiring prior knowledge of the target, specific antibodies, or genetic hypotheses. At Creative Proteomics MassTarget, we deploy an integrated MS-based target identification platform combining chemoproteomics (AP-MS interactomics), ubiquitinomics, quantitative proteomics, and limited proteolysis-mass spectrometry (LiP-MS) to identify the neosubstrate of any molecular glue, confirm its degradation mechanism, and profile selectivity across the proteome. For dedicated interactome mapping of E3 ligase-neosubstrate interactions, our interactomics (AP-MS / proximity labeling) platform provides the affinity purification-MS workflows for unbiased capture of molecular glue-induced protein-protein interactions in cellular lysates and live cells.
Key Advantages:
- Unbiased neosubstrate discovery — no antibody, no genetic hypothesis, no prior target knowledge required.
- Multi-platform orthogonal validation — AP-MS interactomics + ubiquitinomics + quantitative proteomics + LiP-MS under a single project.
- Compatible with all major E3 ligases — established protocols for CRBN, VHL, MDM2, and novel E3 systems.
- Works from limited compound — as little as 1–5 mg of molecular glue sufficient for a full target ID campaign.
- Turnaround: 3–5 weeks depending on platform scope and number of compounds.