Native Metabolomics for Ligand Discovery — Identify Protein-Bound Metabolites Directly from Complex Mixtures
Screen your entire metabolome against a protein target in a single 10-minute LC-MS run — no pre-purification, no immobilization, no labeling.
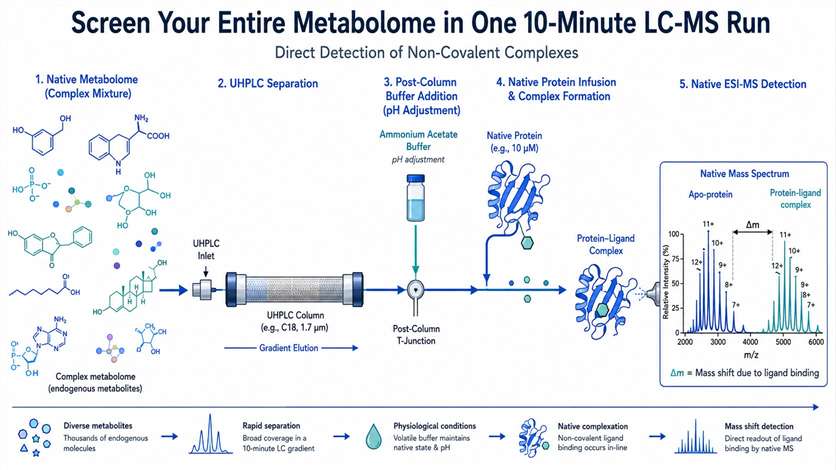
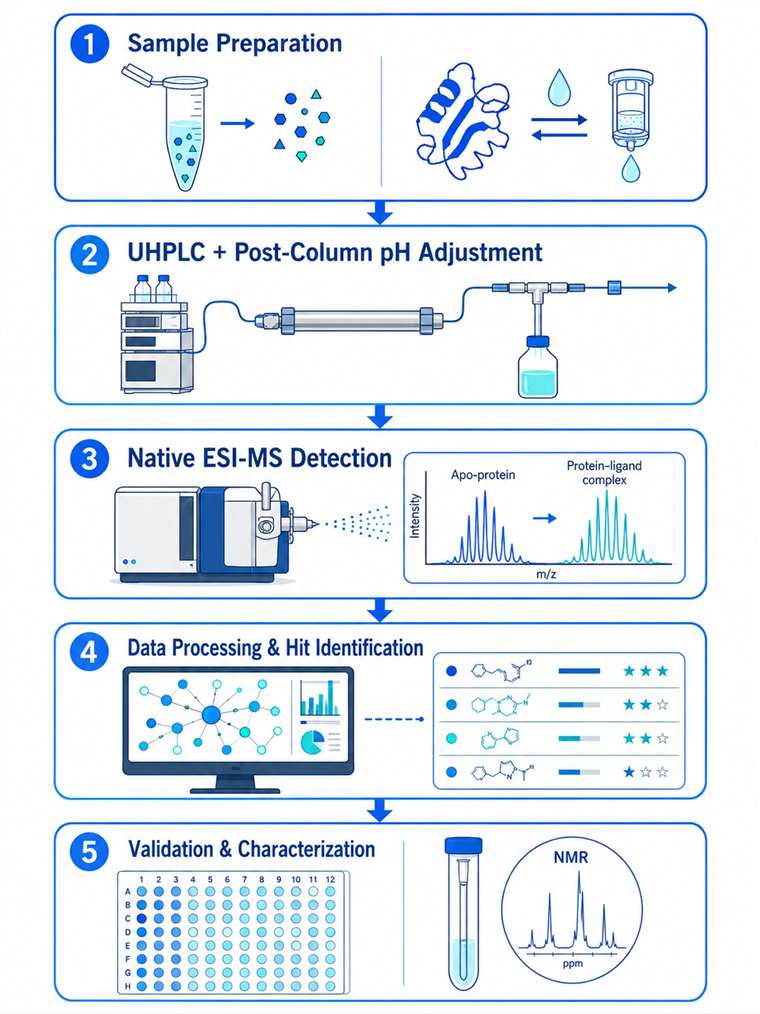
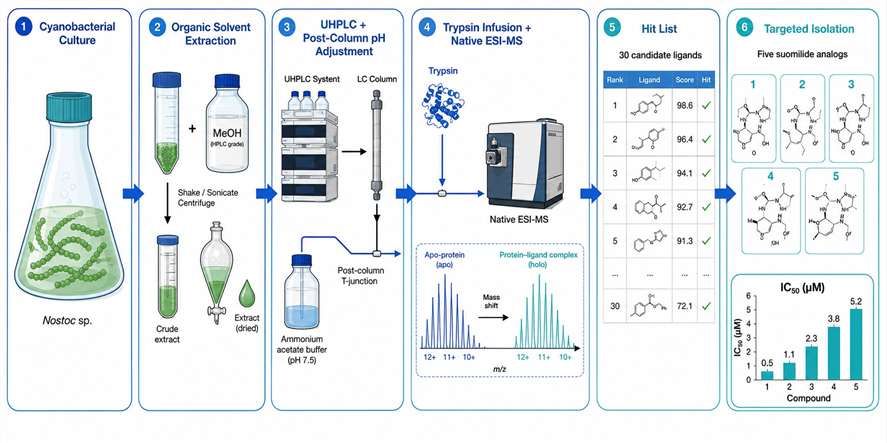
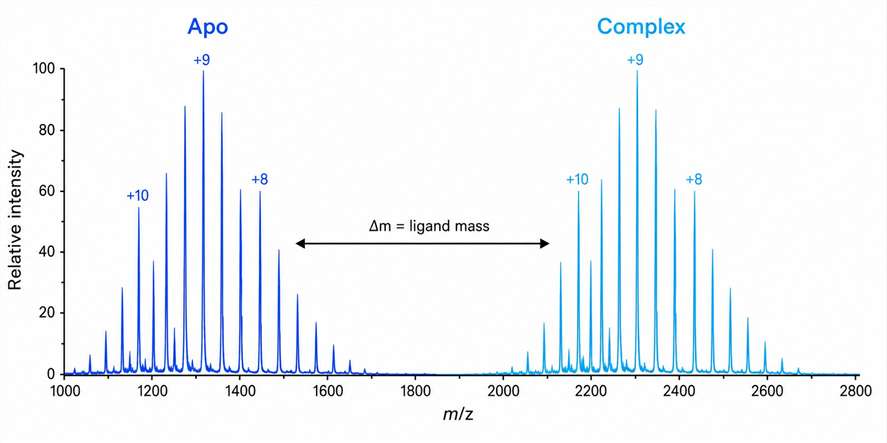
You have a protein target and a complex biological extract. Somewhere in that mixture, a metabolite binds to your protein — but finding it using traditional methods could take months of iterative fractionation. Native metabolomics changes that. We combine untargeted LC-MS/MS metabolomics with native electrospray ionization mass spectrometry to directly detect non-covalent protein-ligand complexes from crude extracts. A single 10-minute LC-MS run screens your entire metabolome against your protein target, identifying bound metabolites by their mass shift and charge state distribution. The result: hit discovery accelerated 10–100× compared to traditional bioassay-guided fractionation, with unambiguous binding evidence.
Key Advantages:
- Screens entire metabolome in a single LC-MS run
- Direct detection of non-covalent protein-ligand complexes
- No target immobilization, labeling, or pre-purification required
- Works with crude extracts, complex mixtures, and diverse protein targets
- Seamless integration with downstream structure elucidation