A machine learning-based chemoproteomic approach to identify drug targets and binding sites in complex proteomes. Nat Commun 11, 4200 (2020). https://www.nature.com/articles/s41467-020-18071-x.pdf
Background
Identifying target proteins for new drugs discovered through phenotypic screening is incredibly difficult when you cannot chemically modify the drug. Traditional label-free methods often suffer from exceptionally high false-positive rates, pointing researchers toward the wrong proteins (typically highly abundant background proteins), and they completely fail to pinpoint where the drug actually binds, leaving medicinal chemists working in the dark.
Methods
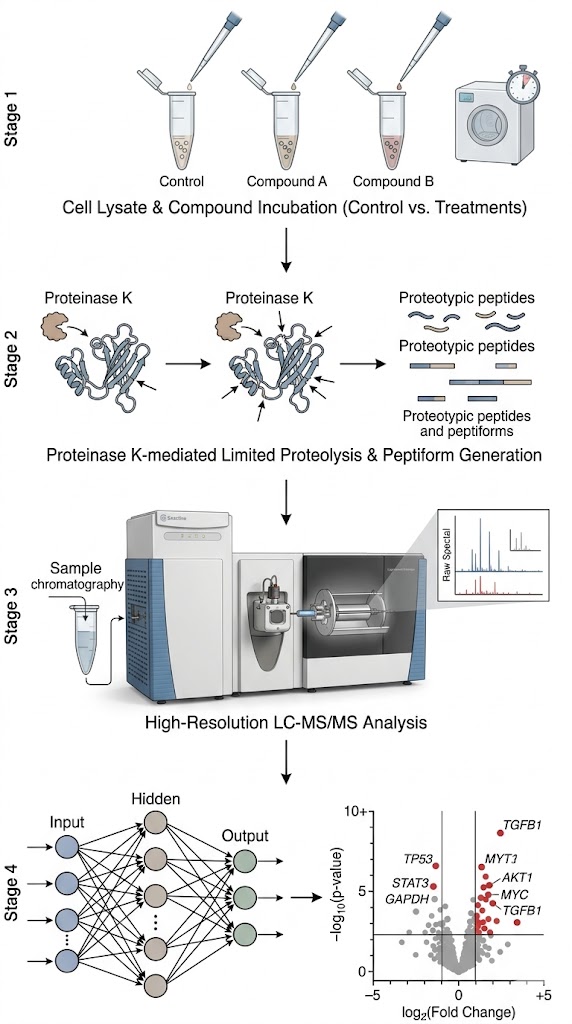
To overcome this, researchers applied the advanced LiP-Quant workflow. They incubated complex human cell lysates with several well-characterized kinase inhibitors across a full concentration gradient. They then performed strictly controlled limited proteolysis with Proteinase K. The resulting peptide mixtures were quantified using high-resolution, data-independent acquisition (DIA) mass spectrometry. Crucially, a novel machine learning (ML) algorithm (trained on hundreds of known protein-ligand interactions) was applied to the massive data array to extract true dose-response signatures, eliminate random structural fluctuations, and calculate specific, high-confidence "LiP scores."
Results
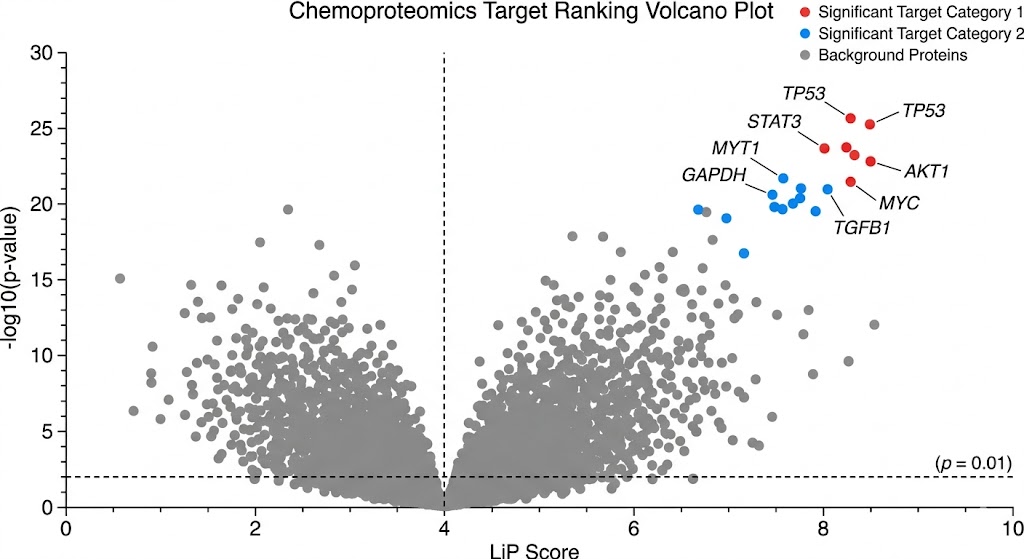
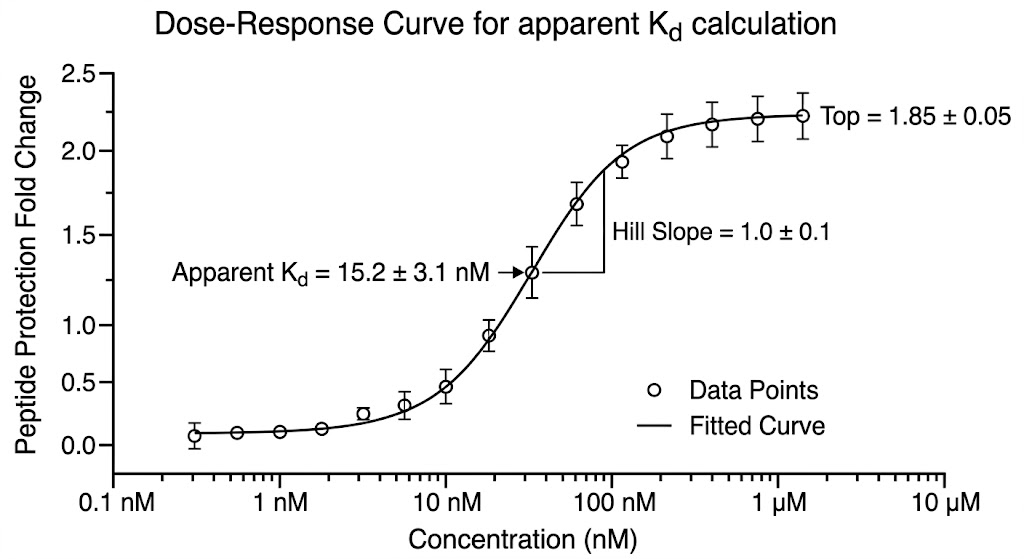
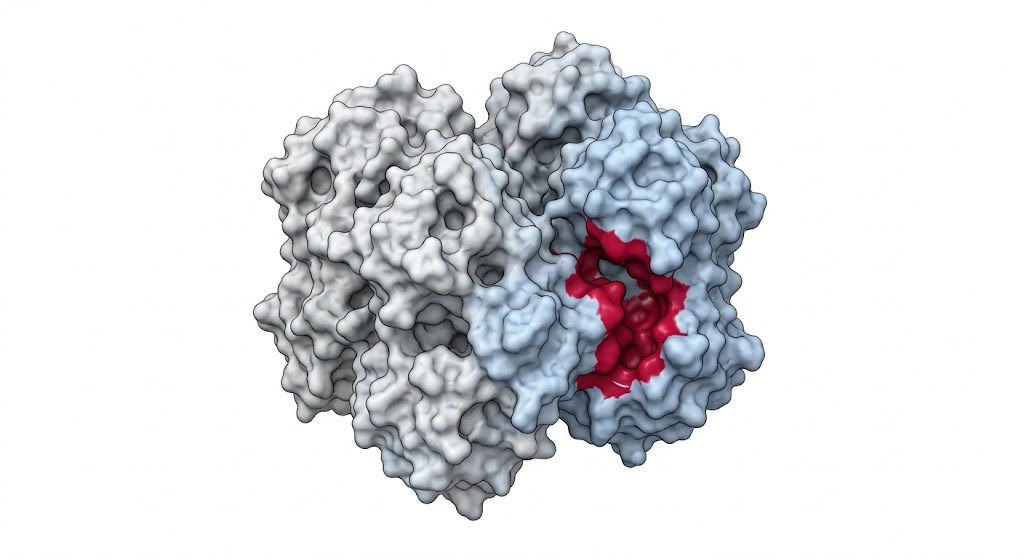
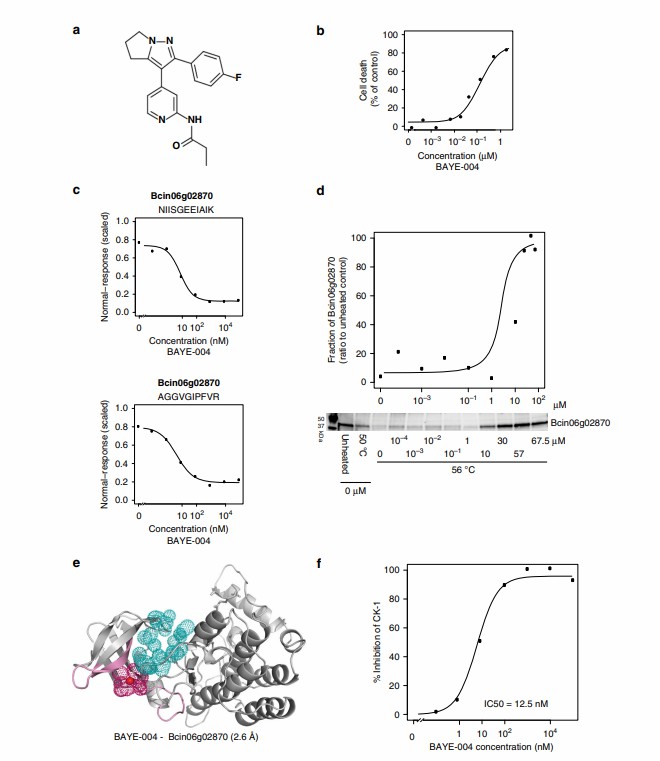
As shown in Figure 4 of the published study, the LiP-Quant platform successfully identified the specific binding targets of the kinase inhibitors. More importantly, the dose-response data accurately determined the apparent Kd values directly within the crude lysate, matching the known biochemical affinities of the drugs. The machine learning model perfectly distinguished the true targets from the high-abundance background proteome. Furthermore, the protected peptides identified by the mass spectrometer successfully mapped the exact ATP-binding pockets on the kinase targets when projected onto 3D protein models.
Conclusion
LiP-Quant, powered by machine learning, is an exceptionally robust label-free platform. It allows drug discovery teams to confidently deconvolve targets from phenotypic screens, comprehensively profile off-target effects across the proteome, and understand the molecular mechanism of action for early-stage drug candidates without the risk of destroying compound activity through chemical tagging.