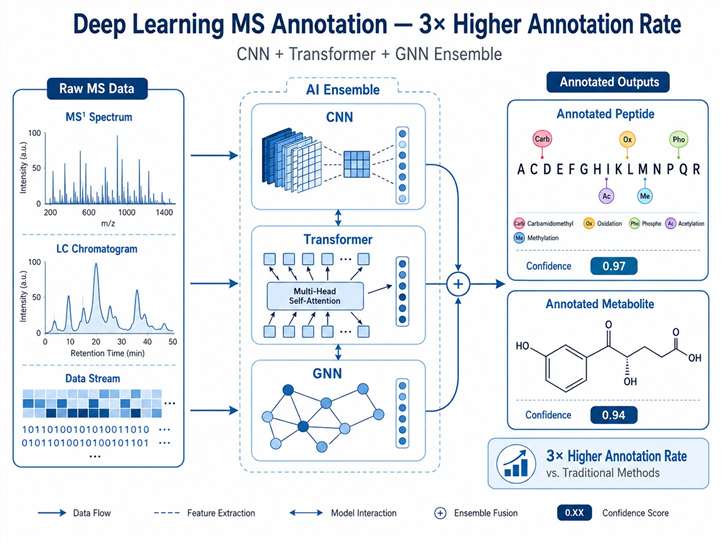
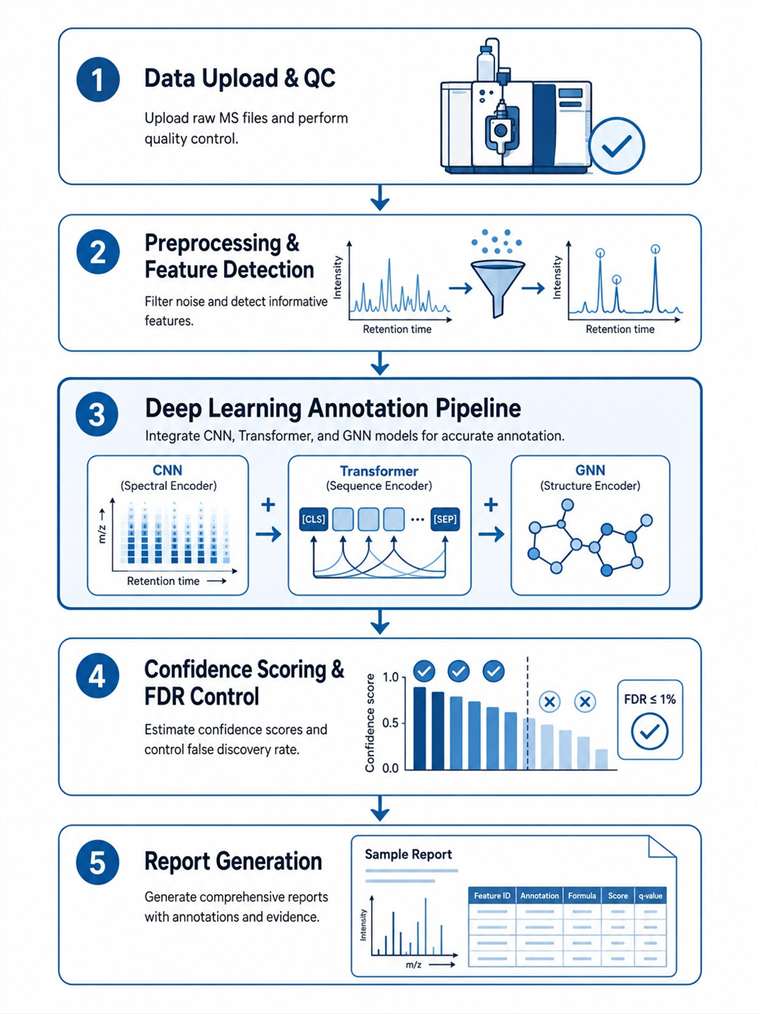
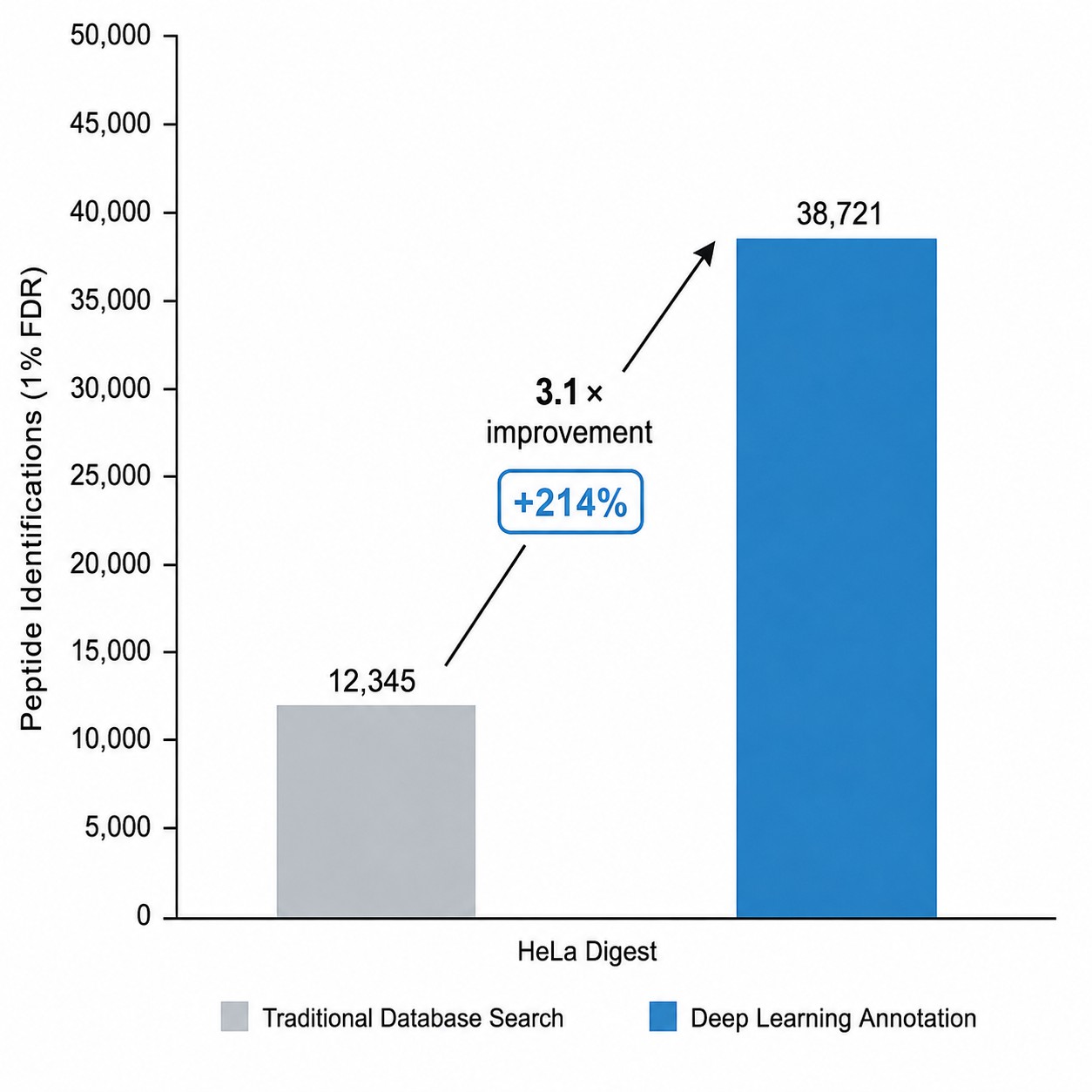
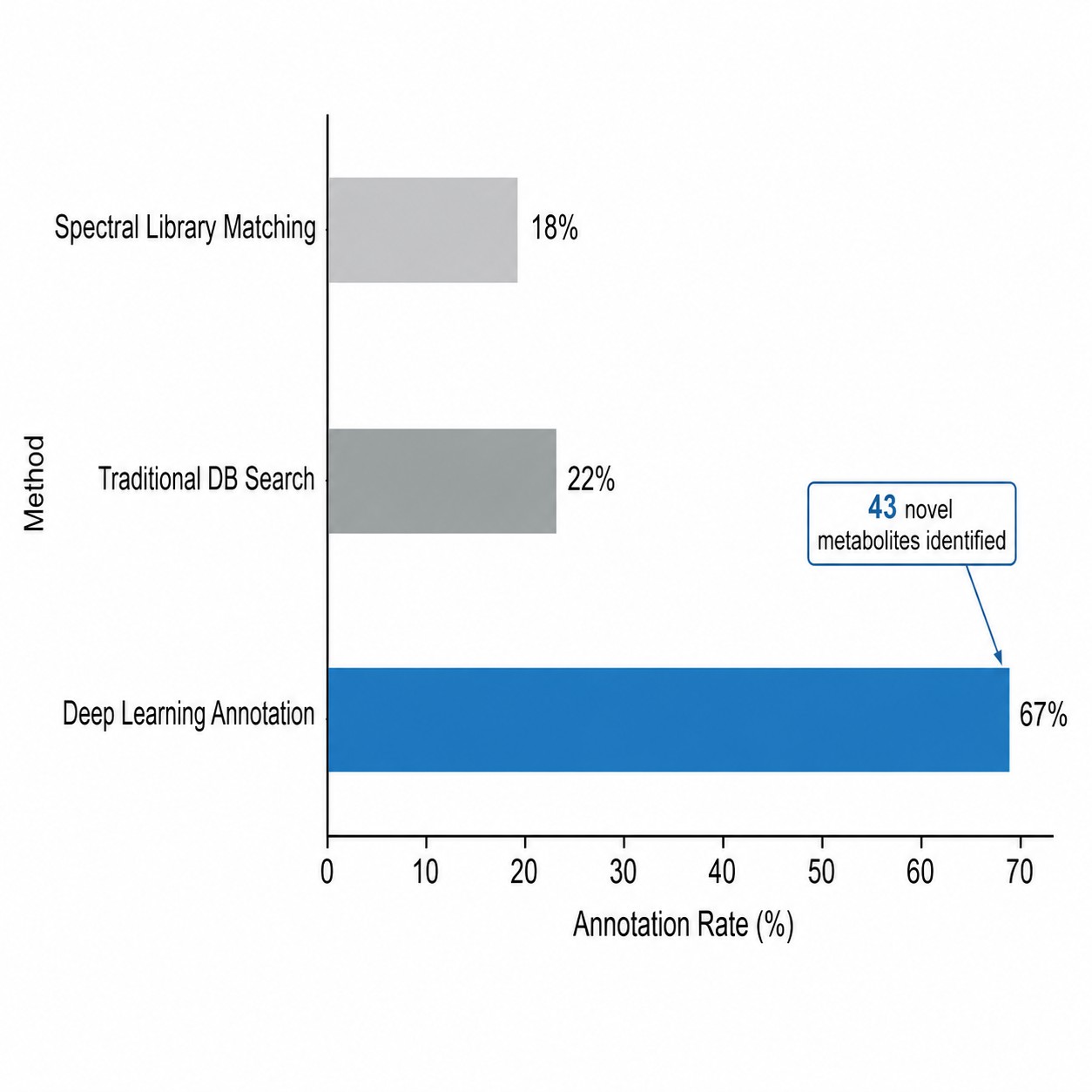
Deep Learning–Assisted MS Annotation — From Raw Spectra to Confident Identifications
We combine CNN, Transformer, and GNN models to push annotation rates 3× beyond what traditional database search can achieve — across both proteomics and metabolomics.
A fully managed service: you upload raw data, we return publication-ready results. Our ensemble deep learning pipeline achieves 90%+ accuracy on known compound libraries with a 48-hour typical turnaround for standard projects.
Key Metrics:
- 3× higher annotation rate vs. traditional database search
- 90%+ accuracy on known compound libraries
- 48-hour typical turnaround for standard projects
- Both proteomics and metabolomics in one platform