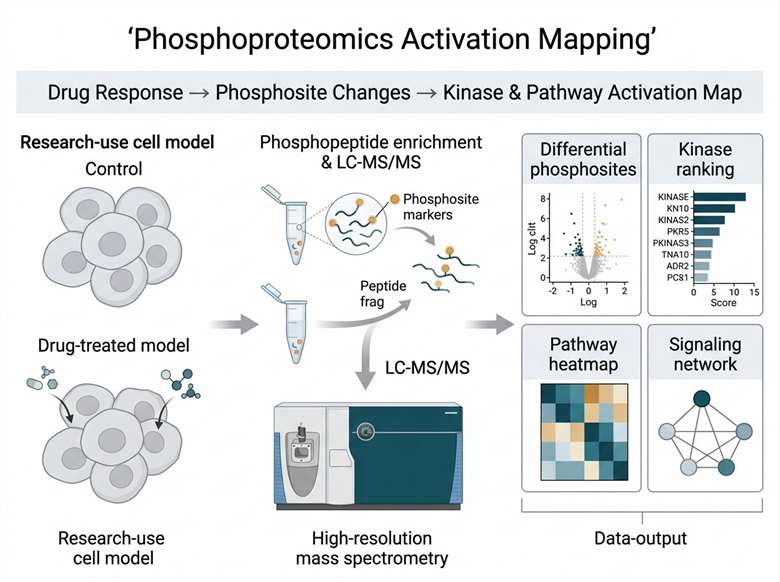
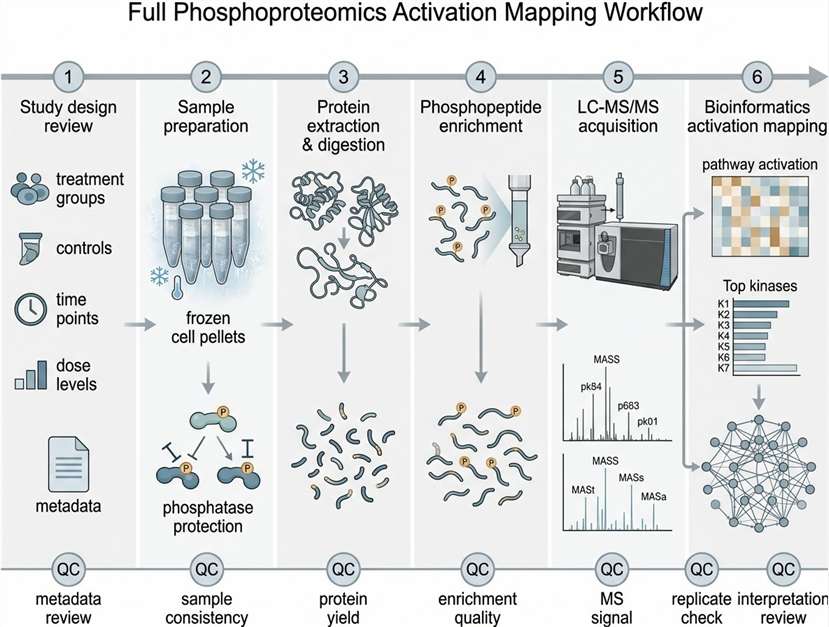
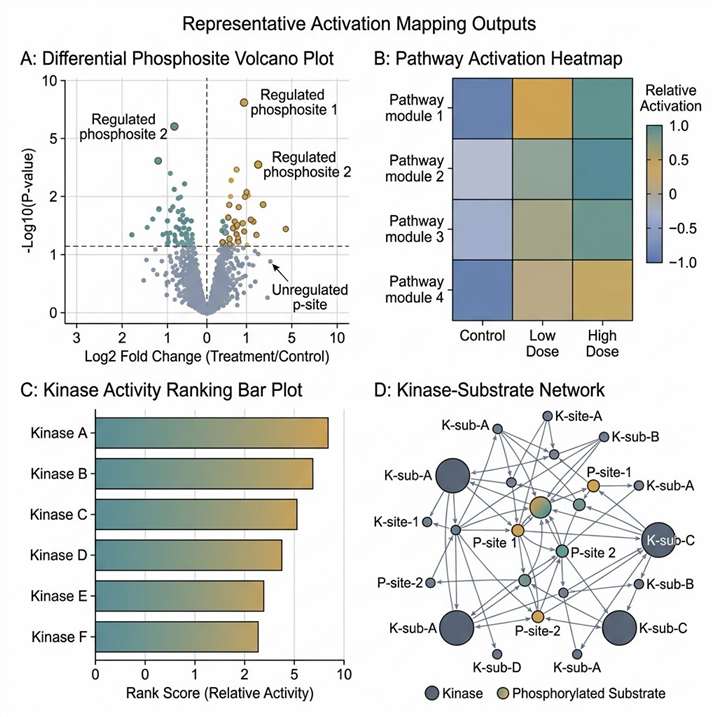
Background
Liang et al. published a 2024 open-access study in Cell Discovery titled CIP2A induces PKM2 tetramer formation and oxidative phosphorylation in non-small cell lung cancer. The study investigated how CIP2A regulates metabolic behavior in non-small cell lung cancer models.
The authors focused on PKM2 because PKM2 can exist in different oligomeric states and those states are linked to different metabolic functions. The central question was how CIP2A affects PKM2 state, phosphorylation, cellular localization, oxidative metabolism, and response to combined treatment conditions.
Methods
The study used multiple experimental approaches across cell models, protein interaction experiments, metabolic assays, phosphorylation analysis, and animal tumor samples. The authors identified PKM2 as a CIP2A-binding protein by immunoprecipitation followed by mass spectrometry. They then used co-immunoprecipitation, pull-down assays, immunofluorescence, proximity ligation assays, gel filtration, crosslinking experiments, site-directed mutants, phospho-specific analysis, Seahorse metabolic assays, and tumor sample staining.
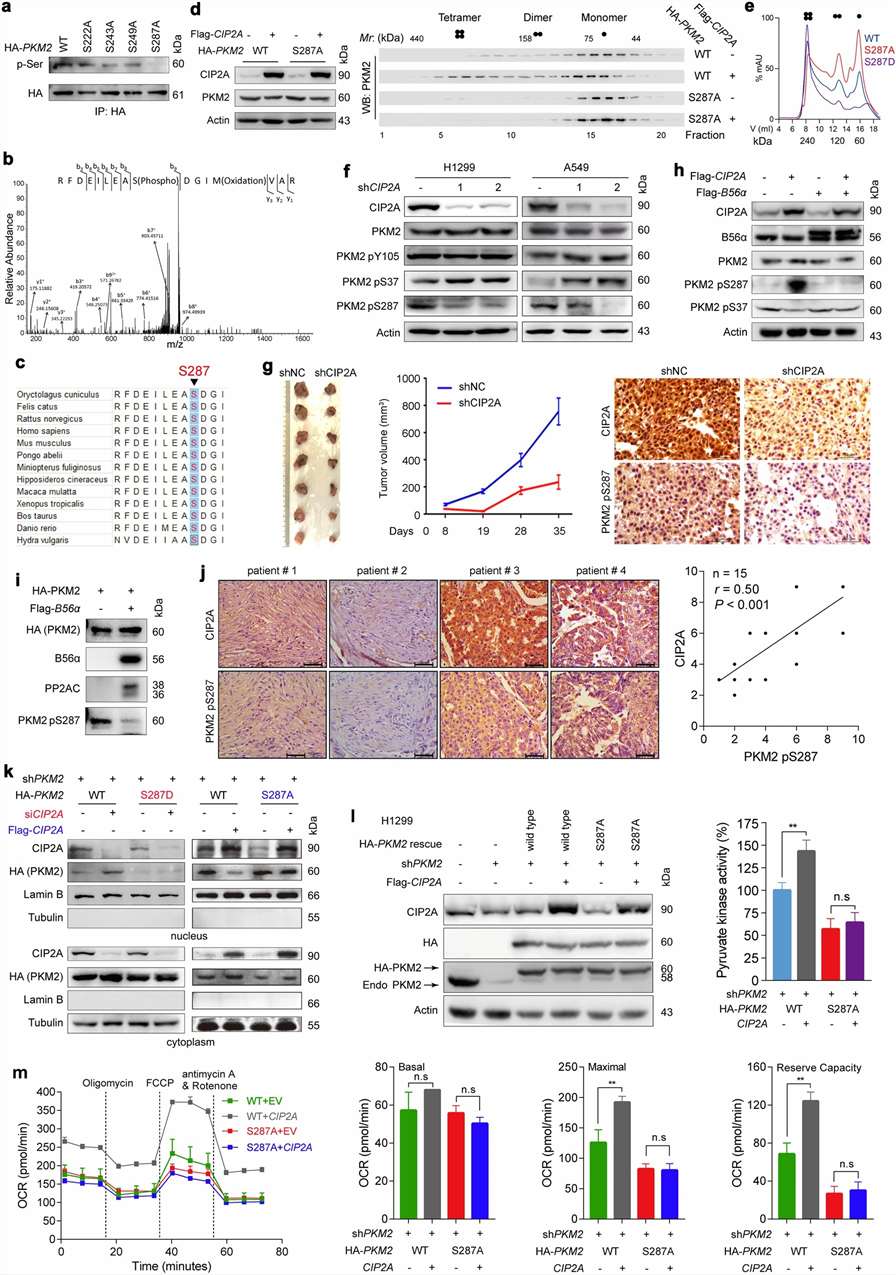
For the activation-mapping angle, Fig. 5 is especially relevant. In this figure, the authors tested whether serine 287 is a critical phosphorylation site of PKM2. They used S287A and S287D mutants, MS-based site evidence, size exclusion chromatography, phosphorylation-specific analyses, pyruvate kinase activity assays, and OCR mitochondrial respiration measurements.
Results
The study reported several connected observations.
First, CIP2A was found to bind PKM2 and promote PKM2 tetramer formation. The authors showed that CIP2A knockdown reduced PKM2 tetramer levels, while CIP2A overexpression shifted PKM2 toward higher-molecular-weight forms.
Second, S287 was identified as a key phosphorylation site. In Fig. 5a, the S287A mutation reduced PKM2 phosphorylation, while the other tested serine-to-alanine mutants did not significantly reduce overall PKM2 phosphorylation under the same condition. In Fig. 5b, MS analysis showed PKM2 S287 phosphorylation. In Fig. 5d and Fig. 5e, PKM2 S287 status was linked to oligomeric state, including tetramer, dimer, and monomer distribution.
Third, the authors connected this phosphorylation site to tumor-model and tissue evidence. In Fig. 5g, CIP2A knockdown inhibited tumor growth and reduced PKM2 S287 phosphorylation in tumor samples from mice inoculated with shCIP2A-expressing A549 cells. In Fig. 5j, IHC staining of 15 human lung adenocarcinoma specimens showed that PKM2 pS287 levels and CIP2A levels were correlated.
Fourth, S287 phosphorylation was linked to functional output. In Fig. 5l, PKM2 S287A showed lower pyruvate kinase activity compared with the relevant control context. In Fig. 5m, OCR mitochondrial respiration parameters were also affected, with measurements reported from three independent experiments. The authors further reported that PKM2 S287 phosphorylation was associated with CIP2A regulation and metabolic state.
Fifth, the study connected phosphorylation state to treatment-response logic. The abstract reported that CIP2A-targeting compounds synergized with a glycolysis inhibitor in suppressing cell proliferation in both in vitro and in vivo models.
Conclusion
This paper is a strong example of why phosphorylation-site evidence matters beyond site identification. The S287 phosphorylation event was not treated as an isolated modification. It was connected to PKM2 oligomeric state, pyruvate kinase activity, mitochondrial respiration, tumor-model observations, and treatment-response behavior.
For phosphoproteomics activation mapping, this is the type of logic we aim to support: a phosphosite change should be interpreted in relation to protein function, pathway activity, biological state, and practical validation plans.