Biomarker to Target Translation by MS — Quantitative Proteomics, Thermal Profiling, and Multi-Omics Integration for Target Discovery and Validation
From correlative biomarker to validated drug target: a single MS-based pipeline bridging discovery proteomics, target deconvolution, and engagement confirmation.
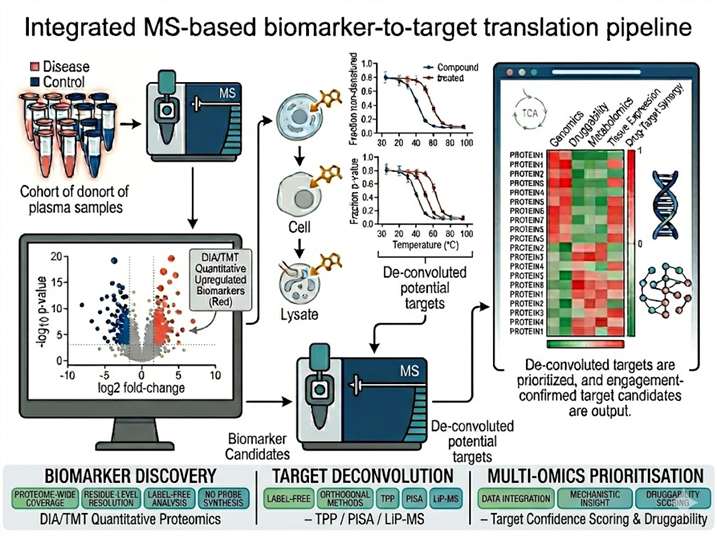
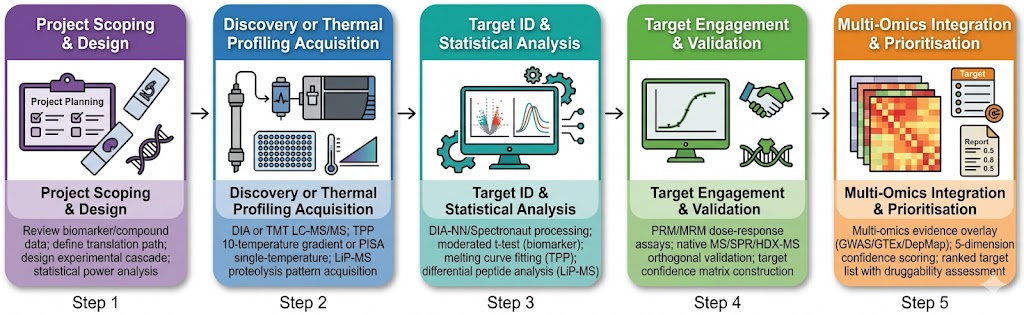
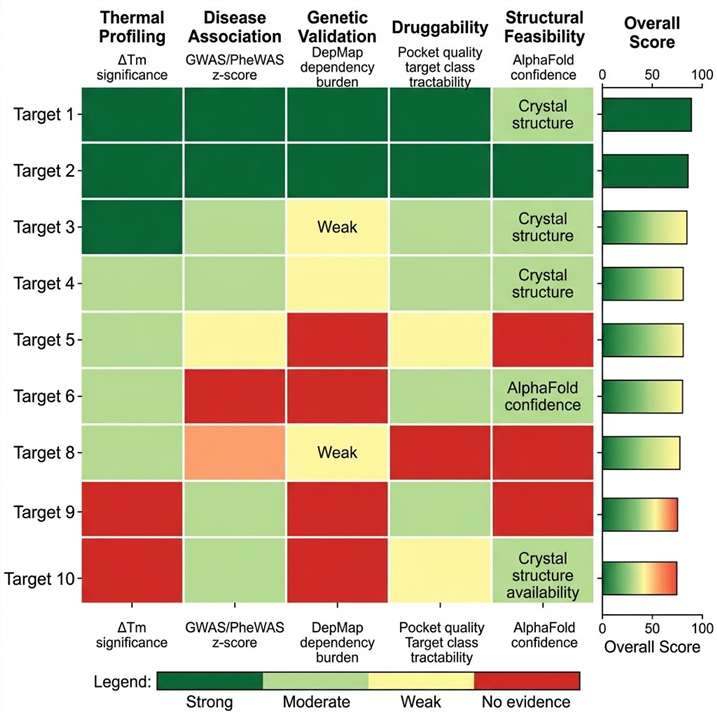
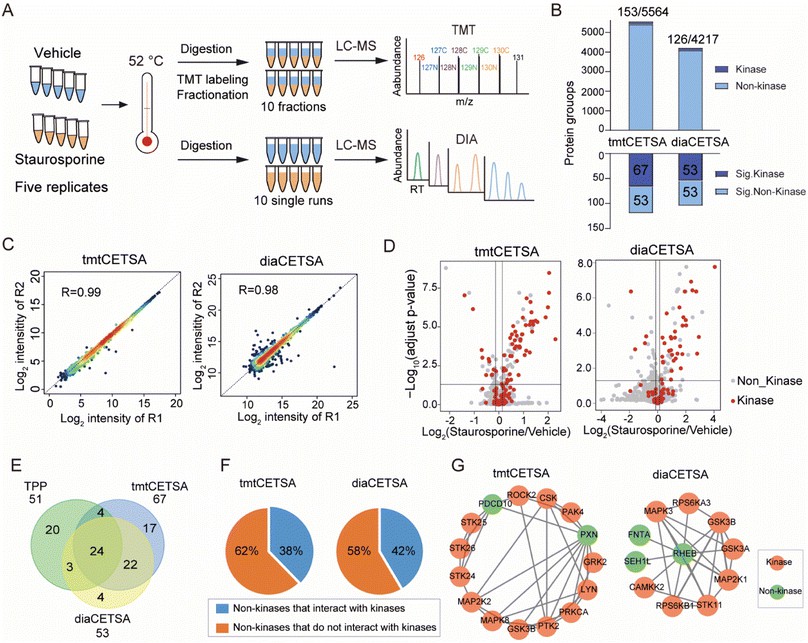
The gap between biomarker discovery and target validation is one of the most resource-intensive transitions in drug discovery. A proteomic biomarker — a protein differentially abundant between disease and control — is a correlation, not a mechanism. Transforming that correlation into a druggable target requires: confirmation that the protein is functionally involved in disease biology, identification of compounds that engage it, quantification of target engagement at proteome scale, and prioritisation of the most therapeutically tractable candidates. Each step demands a different analytical modality, and the fragmentation of these capabilities across separate providers creates data compatibility gaps, sample handover losses, and project timeline delays. Our biomarker-to-target translation service integrates quantitative discovery proteomics, thermal proteome profiling (TPP and PISA), limited proteolysis-MS (LiP-MS), targeted MS assay development, and multi-omics computational integration within a single pipeline — enabling translational scientists to move from biomarker identification to validated, engagement-confirmed target candidates without switching platforms or providers.
Key Advantages:
- End-to-end pipeline from biomarker discovery to target engagement — one platform, one project team, one data framework.
- Proteome-wide target deconvolution by TPP, PISA, and LiP-MS — no target prefamiliarisation or antibody requirement.
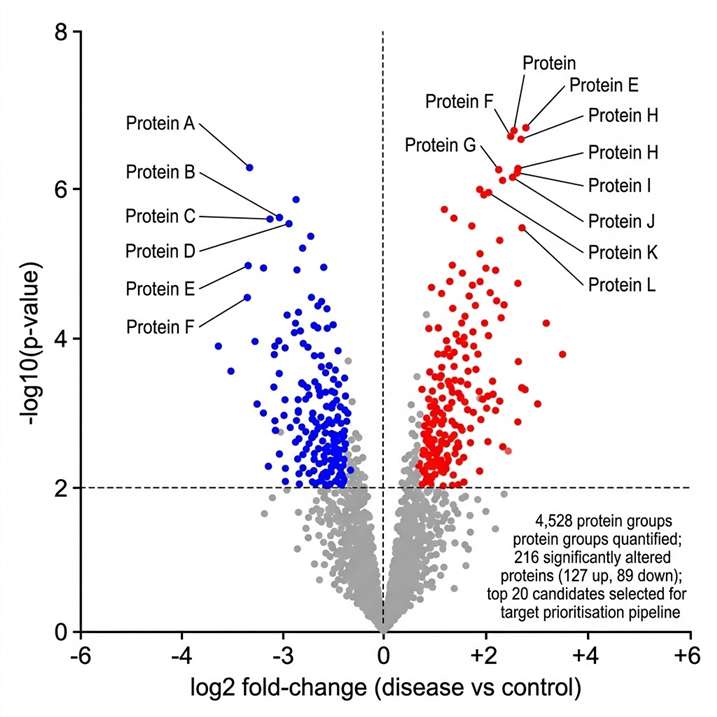
- Quantitative proteomics at cohort scale — DIA and TMT workflows for biomarker discovery across 100s of samples.
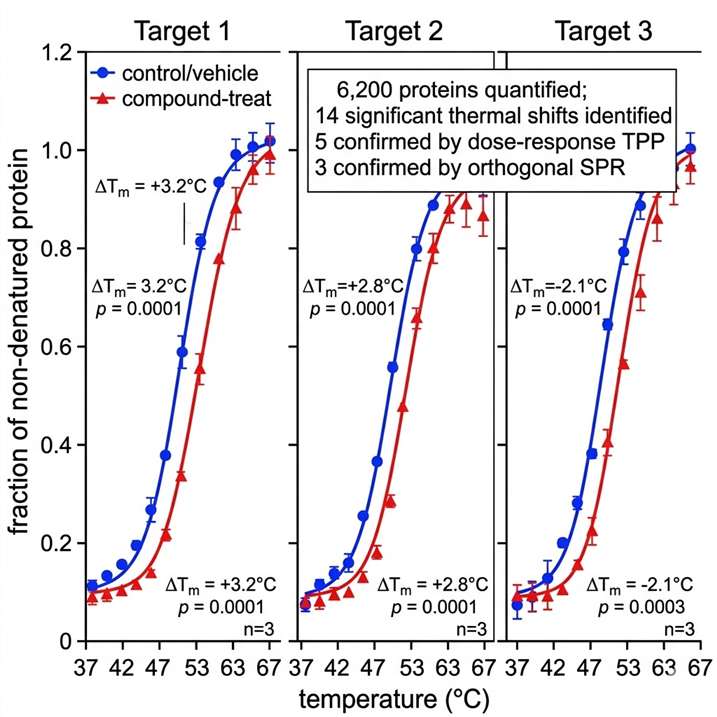
- Target engagement quantification at the proteome level — measure compound-binding events across thousands of proteins simultaneously.
- Multi-omics integration — proteomics, transcriptomics, and metabolomics data fused for target prioritisation with confidence scoring.
- Orthogonal validation cascade — thermal profiling hits confirmed by native MS, HDX-MS, crosslinking MS, or SPR within the same project team.