Pharmaco-Proteomics: MS-Based Drug Response Profiling Service
Translate drug treatment into protein-level evidence — quantitative, pathway-resolved, and decision-ready.
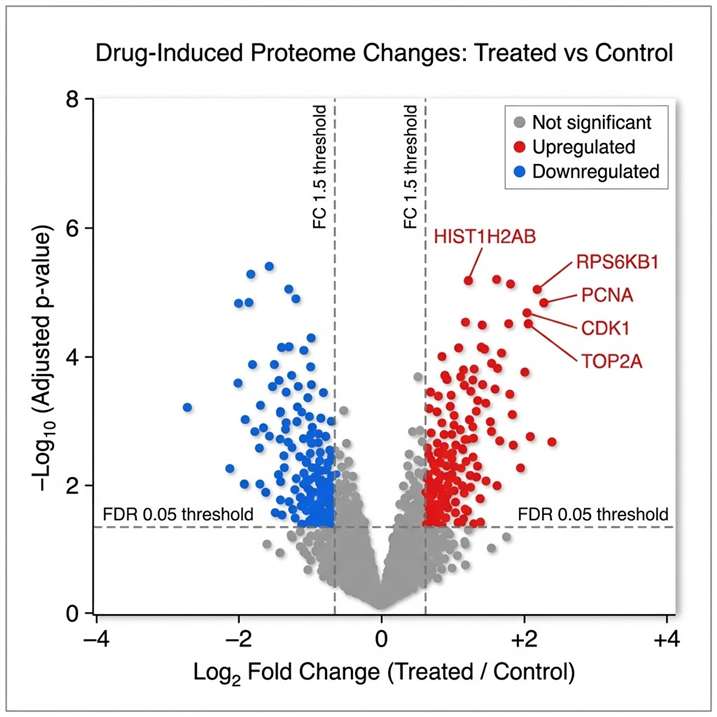
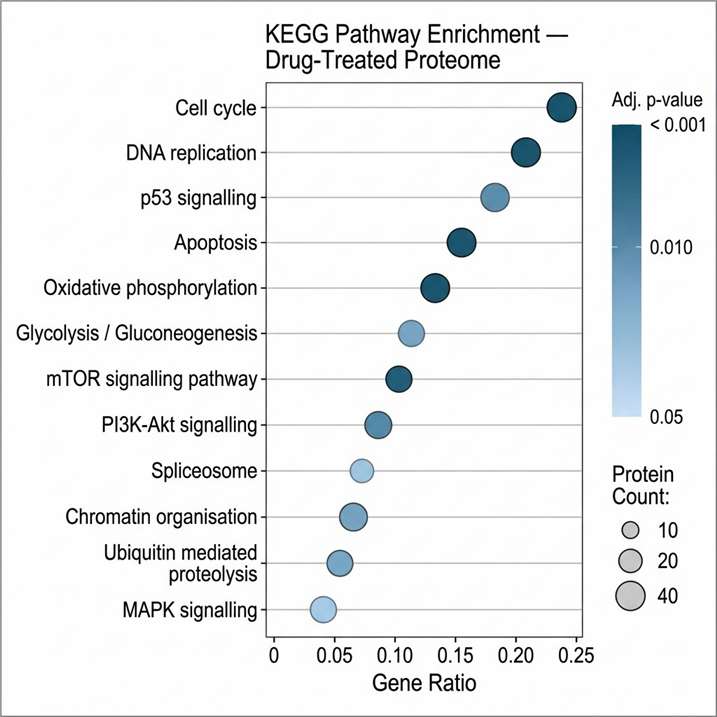
Pharmaco-proteomics applies mass spectrometry-based quantitative proteomics to characterise how a drug or compound reshapes the cellular proteome. Rather than inferring protein-level events from gene expression, it measures thousands of proteins directly — capturing drug-induced changes in abundance, modification, interaction, and pathway activity across treated and control samples.
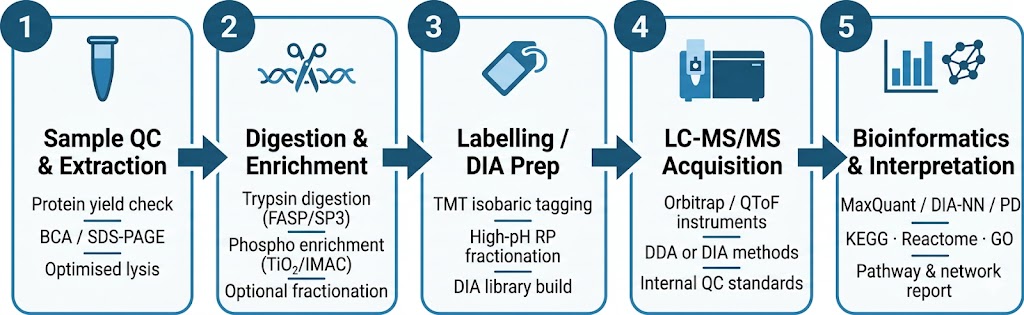
At Creative Proteomics, our pharmaco-proteomics service is built for research teams that need mechanistic depth: MoA elucidation, target engagement confirmation, resistance pathway mapping, and off-target protein change detection — all delivered from a single workflow, with TMT-based quantitative proteomics, label-free quantification, or DIA quantification options.
Key Advantages:
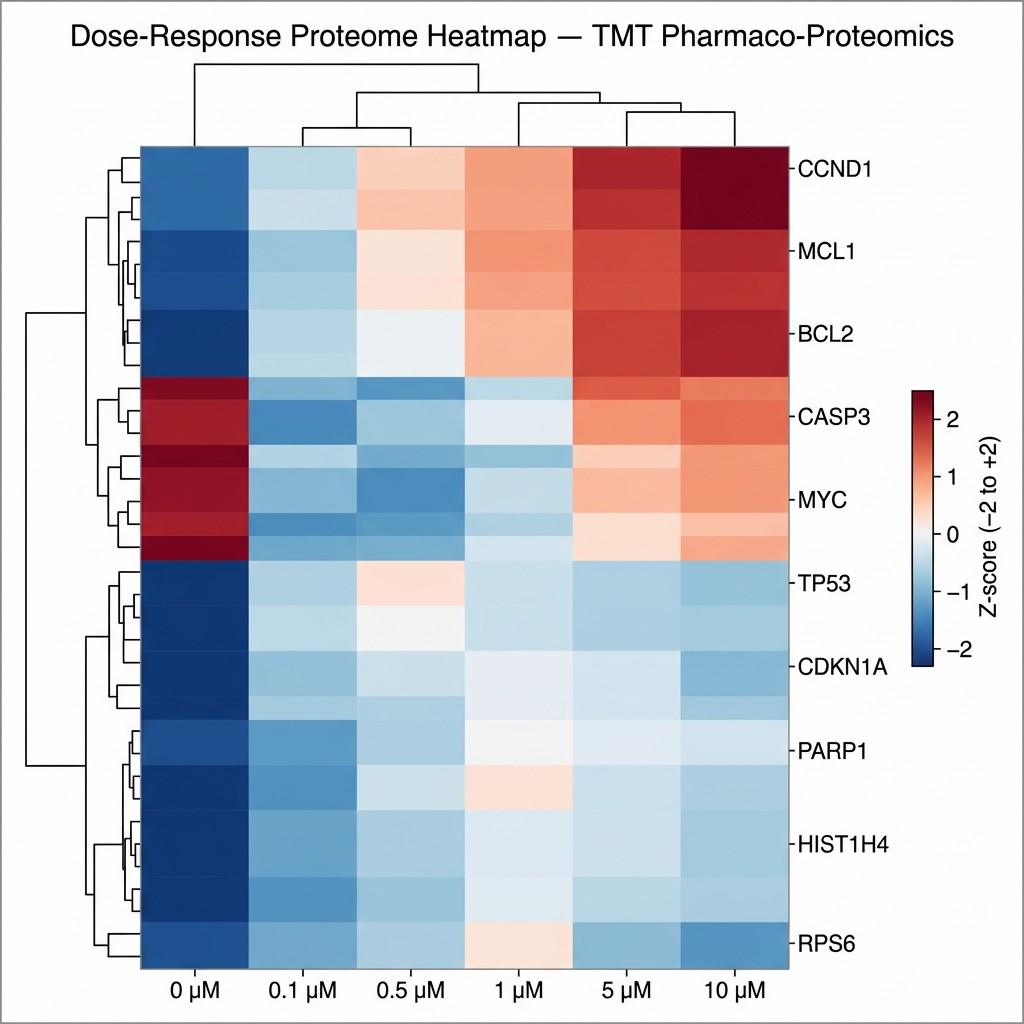
- Comprehensive proteome coverage across drug-treated vs control groups, with multiple quantification strategies (TMT, LFQ, DIA-MS).
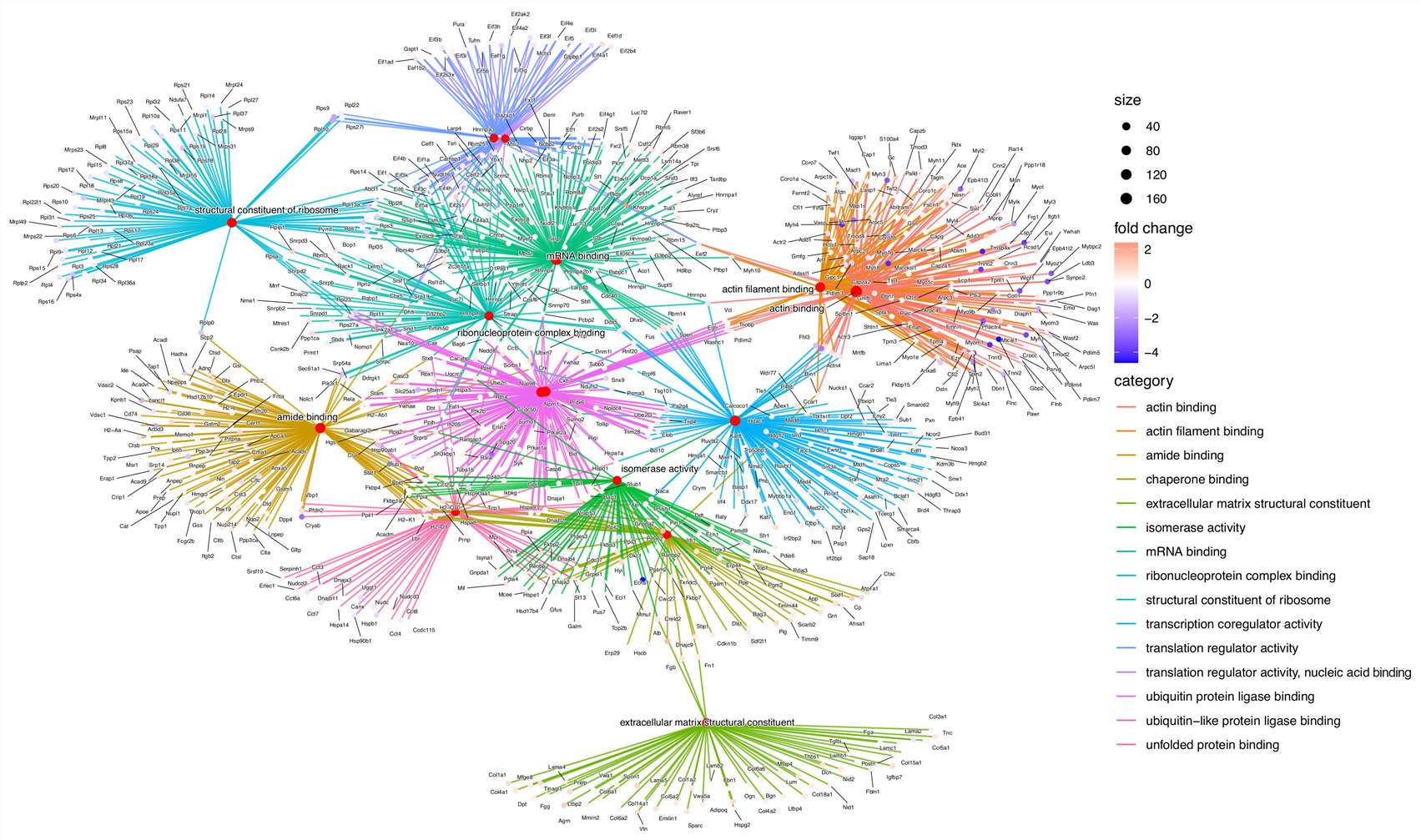
- Pathway and network analysis delivered alongside protein abundance tables — actionable biology, not just data files.
- Compatible with cells, tissues, plasma, and complex biological matrices; flexible sample intake including FFPE.
- Bioinformatics integration covering KEGG, Reactome, and Gene Ontology for mechanistic interpretation.