Richter H., Gover O., Schwartz B. "Anti-Inflammatory Activity of Black Soldier Fly Oil Associated with Modulation of TLR Signaling: A Metabolomic Approach." International Journal of Molecular Sciences 2023, 24(13), 10634. https://doi.org/10.3390/ijms241310634
Research Question
Black soldier fly larvae (BSFL) oil is a sustainable dietary ingredient rich in medium-chain fatty acids, particularly C12:0 (lauric acid). Its anti-inflammatory activity had been observed in cellular and animal models, but the precise metabolic mechanism — specifically how it modulates Toll-like receptor (TLR) signalling and downstream inflammatory metabolite production — was unknown. Researchers used Creative Proteomics' LC-MS-based metabolomics platform to characterise the eicosanoid and oxylipin metabolic landscape in treated macrophage and colitis models, aiming to connect the lipid treatment to a specific TLR-linked MoA.
Methods
THP-1 and J774A.1 macrophage cell lines were activated with TLR4 (LPS) or TLR2 (Pam3CSK4) agonists and treated with BSFL oil or purified C12:0. An in vivo dextran sulfate sodium (DSS)-induced acute colitis mouse model was used for physiological validation. Creative Proteomics provided LC-MS-based quantitative analysis of eicosanoids and oxylipins — a specialised untargeted-to-targeted metabolomics workflow — capturing lipid mediator changes across treatment conditions. Key endpoints measured included proinflammatory cytokine suppression, mTOR signalling activity, and PPAR pathway activation, all cross-referenced with the LC-MS metabolomic readout.
Key Findings
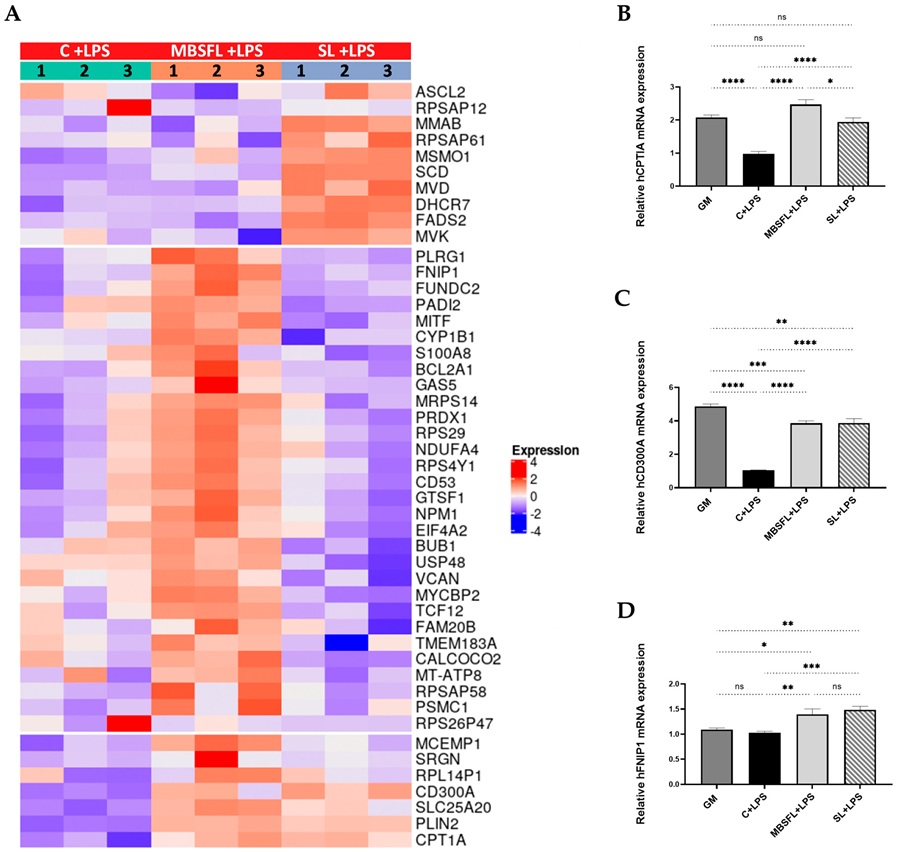
BSFL oil — but not purified C12:0 alone — suppressed proinflammatory cytokine release in Pam3CSK4-stimulated macrophages (TLR2 activation), demonstrating a MoA distinct from simple lauric acid activity. The metabolomic analysis of eicosanoids and oxylipins identified a lipid mediator signature consistent with modulation of TLR2-linked inflammatory signalling, including perturbation of mTOR-dependent metabolic reprogramming and activation of PPAR-related anti-inflammatory metabolic pathways. In the DSS colitis mouse model, BSFL oil treatment produced measurable protective effects on colon tissue integrity, with the metabolic fingerprint providing mechanistic context for the observed anti-inflammatory phenotype that would not have been accessible from cytokine assays or gene expression alone.
Significance for Untargeted Metabolomics MoA Research
This study illustrates the core value of metabolomics-based MoA characterisation: the activity difference between BSFL oil and its purified major fatty acid (C12:0) was only mechanistically interpretable through the lipid mediator metabolic landscape. The eicosanoid and oxylipin profile revealed TLR2-specific pathway engagement that a protein-level or transcript-level assay alone would not have resolved. The study demonstrates how LC-MS-based metabolomics translates a biological activity phenotype into a specific pathway-level MoA fingerprint — exactly the evidence generation pathway that Creative Proteomics' untargeted metabolomics MoA service is designed to support.