Every protein interaction map starts with a capture decision. Two affinity-based strategies dominate: antibody-mediated co-immunoprecipitation (Co-IP) and glutathione S-transferase (GST) fusion protein pull-down. Both isolate protein complexes from lysate, yet they serve different purposes at different project stages.
Co-IP pulls endogenous complexes from their native cellular environment using an antibody against the bait protein — or an epitope tag when no suitable antibody exists. The result reflects interactions present in the cell at lysis. GST pull-down uses a recombinant GST-fusion bait expressed in Escherichia coli, immobilized on glutathione-agarose beads, to fish for interacting partners from a separately prepared lysate — a reductionist approach that trades cellular context for biochemical control.
The practical question is not "which method is better" but "which method answers my question now." Validating a candidate from a proximity-labeling screen demands a different tool than de novo mapping of an uncharacterized protein.

Antibody-Based Co-Immunoprecipitation: Antibody Selection and Lysis Optimization
Antibody Selection and IP Validation
The antibody is the most consequential reagent in Co-IP. An antibody giving clean Western blot signal against denatured protein may fail in IP, where it must recognize the native folded epitope — potentially buried in a complex.
Polyclonal antibodies target multiple epitopes, increasing the chance that some remain accessible in the native complex, at the cost of lot variability and higher background. Monoclonal antibodies offer reproducible specificity but recognize a single epitope, making them vulnerable to masking. When an IP-validated antibody is unavailable, those validated for IHC or flow cytometry are the next best candidates — both require native epitope recognition.
Titrate the antibody to find the minimum amount that captures the bait quantitatively. A starting range of 1–10 ug per 500 ug to 1 mg of total lysate protein is reasonable. Excess antibody increases heavy-chain co-elution (~50 kDa) and nonspecific background. Include an isotype-matched IgG control at the same concentration. A knockout lysate provides the most rigorous negative control.
Lysis and Wash Optimization
The lysis buffer must solubilize the compartment containing the bait without stripping away physiological interactors. For cytoplasmic and membrane proteins, 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, and 1% NP-40 or Triton X-100 is the standard starting point. Both are non-ionic detergents that solubilize membranes without denaturing proteins.
RIPA buffer adds 0.5% sodium deoxycholate and 0.1% SDS for nuclear and cytoskeletal proteins, but SDS can dissociate genuine interactors. Systematic comparisons of lysis conditions for AP-MS have shown that detergent choice alone can shift the identified interactome by 30–40%, with SDS-containing buffers selectively losing weaker but biologically relevant interactions [6]. If RIPA is unavoidable, validate by comparing RIPA and NP-40 extracts in a pilot experiment.
Fresh protease inhibitor cocktail, 1 mM PMSF, and phosphatase inhibitors are non-negotiable. Pre-clear lysate with control beads for 30–60 minutes at 4°C before adding IP antibody — a low-effort step that consistently reduces background.
Wash stringency is tuned via salt and detergent. NaCl at 150 mM is standard; 300–500 mM strips electrostatic background but may remove low-affinity partners. Run a pilot salt gradient and identify the highest concentration that retains a known positive interactor. For challenging baits — membrane proteins, low-abundance targets — co-immunoprecipitation services with pre-optimized protocols can accelerate discovery timelines.
Elution Strategies for Co-IP
The choice of elution method determines what ends up in the final sample — and whether it is MS-compatible. Three strategies dominate:
| Method | Mechanism | MS-Compatible? | Best For |
|---|---|---|---|
| SDS boiling (95°C, 5–10 min in Laemmli buffer) | Denatures the antibody-antigen complex; releases everything | No — SDS and antibody chains contaminate | Western blot readout only |
| Low-pH glycine (0.1–0.2 M, pH 2.0–3.0) | Protonates charged residues, disrupts non-covalent interactions | Yes — after neutralization with 1 M Tris pH 8.0 | Native protein recovery for enzymatic assays or MS |
| Competitive peptide (e.g., 3×FLAG peptide at 100–200 ug/mL) | Competes for antibody binding site; displaces tagged bait | Yes — cleanest eluate, no antibody co-elution | Quantitative Co-IP-MS; the gold standard when using epitope-tagged baits |
SDS boiling is the default for Co-IP-Western blot workflows but introduces antibody heavy and light chain contamination (see Antibody Immobilization Strategies below). Low-pH glycine elution preserves native protein conformation but may not fully dissociate high-affinity antibody-antigen pairs. Competitive peptide elution, when available (FLAG, HA, Myc systems), provides the cleanest sample for downstream mass spectrometry.
GST Pull-Down Workflow
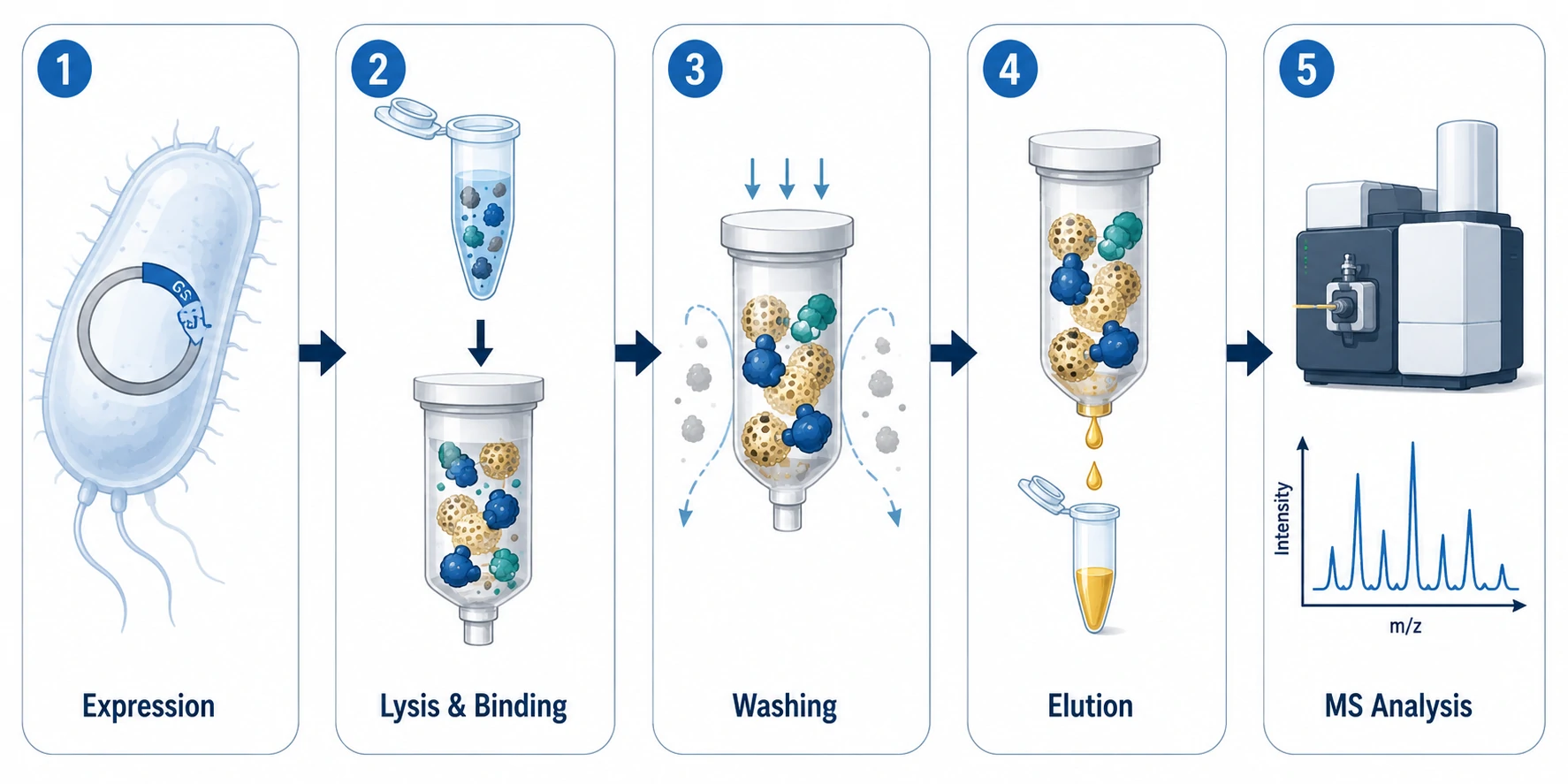
GST Tag Properties and Design
GST is a 26 kDa enzyme from Schistosoma japonicum that binds glutathione with high affinity (Kd ~0.1–1 uM) [1]. As a fusion tag it provides a one-step affinity handle and often improves recombinant protein solubility in E. coli.
Placement matters. N-terminal fusions are standard, but if the bait's N-terminus harbors a signal peptide or functional domain, move GST to the C-terminus or insert a flexible Gly-Ser linker (e.g., (G4S)2) between tag and bait to provide conformational freedom.
When the 26 kDa GST tag is suspected of sterically interfering with bait-prey binding, the tag can be removed by site-specific proteolysis before the pull-down. PreScission protease (a GST-fused 3C protease) and thrombin are the most commonly used enzymes, with their cleavage sites engineered between GST and the bait during cloning. After GST-mediated purification on glutathione-agarose, the column is washed with cleavage buffer and incubated with the protease overnight at 4°C. The cleaved bait — now free of the GST moiety — is collected in the flow-through while GST and the protease remain bound to the column. TEV protease is an alternative when its recognition sequence (ENLYFQ/G) is compatible with the bait's N-terminus. Post-cleavage, the bait should be checked by SDS-PAGE to confirm complete tag removal before proceeding to the pull-down incubation.
GST-Fusion Expression in E. coli
Insolubility in inclusion bodies is the most common failure mode. Lower induction temperature first: shifting from 37°C to 18–25°C slows translation, aiding folding. IPTG can often be reduced to 0.1–0.5 mM. Test multiple strains: BL21(DE3) as standard, BL21(DE3)pLysS for toxic baits, Rosetta or ArcticExpress for codon bias and low-temperature work. After lysis by sonication or French press in PBS with 1% Triton X-100, 1 mM DTT, and protease inhibitors, incubate the soluble fraction with glutathione-agarose beads. For labs that need purified recombinant bait proteins without setting up bacterial expression, Creative Proteomics provides customized synthesized peptide and protein services including tagged constructs ready for pull-down experiments.
Two E. coli contaminants co-purify with GST fusions: DnaK (~70 kDa) and GroEL (~60 kDa) [5]. Wash beads with buffer containing 5 mM ATP and 10 mM MgCl2 after binding to displace DnaK. Always include a GST-alone control — it reveals which bands are GST-binding contaminants rather than bait-specific interactors.
Glutathione Affinity and Elution
Bind GST-fusion protein to glutathione-agarose at 4°C for 1–2 hours with gentle rotation. Wash 3–5 times (10 bed volumes each), then elute with 10–50 mM reduced glutathione in 50 mM Tris-HCl (pH 8.0). Competitive elution is gentler than SDS boiling and releases the complex in a native state suitable for downstream assays or MS. For labs seeking to outsource interaction screening, Creative Proteomics offers validated pull-down assay services with MS-based identification.
Two configurations exist: on-bead (GST-bait remains immobilized, prey lysate added) and post-elution (bait eluted first, incubated with prey in solution, re-captured). On-bead is simpler and sufficient for most interactions; post-elution rescues cases where bead immobilization sterically compromises the bait.
GST vs Antibody-Based Capture: A Side-by-Side Comparison
Choosing between GST pull-down and antibody-based Co-IP is a multi-factor decision. The table below provides a head-to-head comparison across the axes that matter in practice.
| Factor | Antibody Co-IP | GST Pull-Down |
|---|---|---|
| Bait source | Endogenous from any cell type | Recombinant from E. coli (requires cloning) |
| Bait conformation | Native, with physiological PTMs | May lack eukaryotic PTMs; risk of misfolding |
| Scalability | Limited by antibody cost and lysate volume | High — liters of bacterial culture |
| Specificity | Depends on antibody quality; higher background | GST-alone control cleanly subtracts nonspecific binders |
| Capture efficiency | Variable (antibody affinity, epitope accessibility) | High (GST-glutathione affinity is robust) |
| Throughput | Low to moderate | Moderate to high |
| Cost per experiment | High (antibody is the dominant cost) | Low after cloning (beads and glutathione are inexpensive) |
| Endogenous interactor context | Yes — complexes form in the native cell | No — bait and prey meet on beads |
| Best for | Validating endogenous complexes; studying PTM-dependent interactions | Screening for direct binding partners; mapping domain requirements; producing material for MS |
For most projects, the two methods are complementary rather than competitive. A common discovery-to-validation pipeline uses GST pull-down to screen for candidate interactors, followed by Co-IP in the physiologically relevant cell type to confirm that the interaction occurs endogenously.
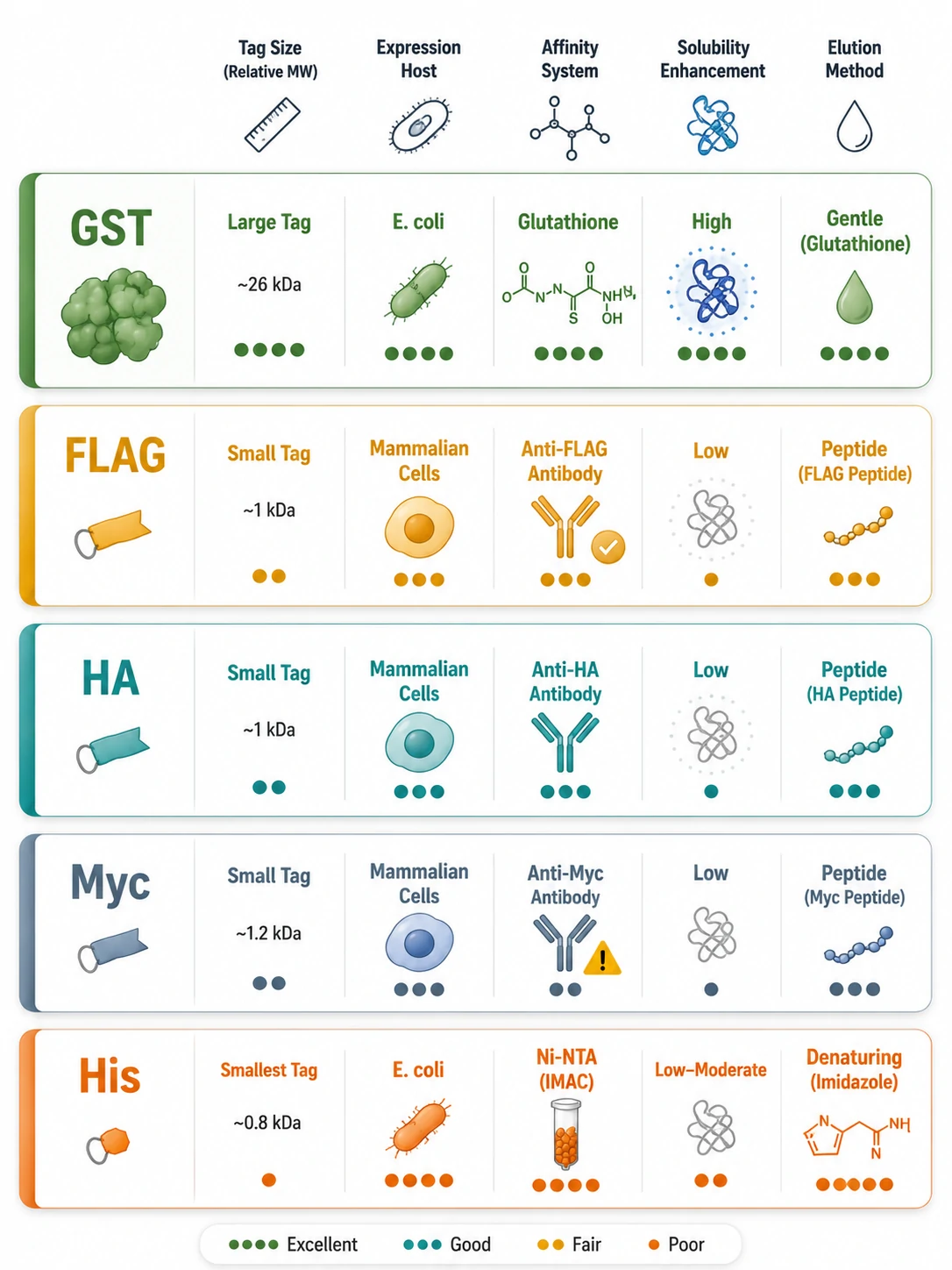
Other Fusion Tags for Interactome Capture
Beyond GST, several epitope and affinity tags serve interactome capture, each with distinct trade-offs in size, antibody availability, and MS compatibility.
FLAG (DYKDDDDK, ~1 kDa) is the top choice for quantitative Co-IP-MS. The M2 monoclonal antibody binds with high specificity at either terminus, and 3×FLAG peptide elution yields the cleanest eluate — no antibody chains, no detergent, no boiling. Anti-FLAG resin is expensive but justified when background must be minimized.
HA (YPYDVPDYA, ~1 kDa) and Myc (EQKLISEEDL, ~1.2 kDa) are small, well-validated tags for mammalian Co-IP. Neither has a direct affinity resin — capture is antibody-mediated — but their compact size minimizes steric interference. HA.11/12CA5 and 9E10 antibodies are reliable workhorses.
GFP (~27 kDa) enables live-cell imaging before lysis, and GFP-Trap nanobody-conjugated beads provide fast, high-affinity capture. The tag's size can sterically block some interactions, but the ability to confirm localization by microscopy before IP is a practical advantage many labs value.
His (6×His, ~0.8 kDa) is the smallest affinity tag and the most economical (Ni-NTA/Co-NTA resin), but high nonspecific background from host metalloproteins limits its standalone use in interactomics. It excels in tandem strategies — His-FLAG dual-tag constructs with sequential Ni-NTA and anti-FLAG purifications approach homogeneous complex preparations. For high-specificity interactome discovery, tandem affinity purification (TAP)-MS combines sequential purification steps to minimize background contaminants.
Tag Selection Decision Matrix
| Experimental Goal | Recommended Tag Strategy | Rationale |
|---|---|---|
| In vitro binding assay with purified proteins | GST on bait | High yield from E. coli; glutathione elution is gentle and specific |
| Endogenous complex validation in mammalian cells | FLAG or HA knock-in, or endogenous antibody Co-IP | Small tag minimizes perturbation; peptide elution for MS compatibility |
| Quantitative Co-IP-MS (discovery) | FLAG or FLAG-HA tandem tag | Peptide elution avoids antibody chain contamination; tandem tags reduce background |
| Screening a panel of prey proteins | GST on bait, lysate-based pull-down | Scalable bait production; cost-effective for many conditions |
| Live-cell imaging + IP | GFP | Confirm localization before lysis; GFP-Trap nanobodies enable fast capture |
| Difficult-to-express eukaryotic bait | HA or Myc in mammalian cells | Avoids E. coli folding problems; bait expressed with native PTMs |
Antibody Immobilization Strategies
Covalent Cross-Linking of Antibodies to Beads
Before cross-linking, selecting the correct bead matrix for antibody binding is essential. Protein A binds strongly to rabbit IgG and human IgG1/IgG2 but poorly to mouse IgG1 and goat IgG. Protein G has broader species coverage — it binds strongly to mouse IgG1, rat IgG, goat IgG, and most mammalian subclasses. When the antibody isotype is unknown or from an uncommon species, a Protein A/G fusion matrix provides the broadest capture range. For rabbit polyclonal antibodies — the most common choice for Co-IP — Protein A alone is usually sufficient and more economical.
When an antibody is simply incubated with Protein A/G beads and then used for IP, the antibody co-elutes with the antigen during SDS boiling — producing the familiar heavy-chain (~50 kDa) and light-chain (~25 kDa) bands on Western blots. Covalently cross-linking the antibody to the beads before IP eliminates this problem: the antibody stays on the beads during elution, and the eluate contains only the bait and its interacting partners.
The standard reagent is dimethyl pimelimidate (DMP), a homobifunctional imidoester that cross-links lysine side chains to form amidine bonds. Protocol: bind antibody to Protein A/G beads, wash, incubate with 5–20 mM DMP in 0.1–0.2 M triethanolamine (pH 8.2) for 30–60 minutes, quench with 50 mM ethanolamine or Tris. BS3, an NHS-ester with a longer spacer (1.14 vs 0.9 nm), is a water-soluble alternative when lysine spacing is suboptimal. Verify cross-linking by boiling a bead aliquot on SDS-PAGE — a properly cross-linked antibody releases no chains.
Light and Heavy Chain Interference
In standard Co-IP-Western blot, SDS-PAGE sample buffer reduces antibody disulfide bonds, releasing heavy chain (~50 kDa) and light chain (~25 kDa) into the eluate. If bait or prey runs near either mass, antibody bands mask the target.
Four solutions, in order of preference: (1) Cross-link antibody to beads — eliminates chains at the source. (2) Use conformation-specific secondary antibodies (TrueBlot) that recognize native IgG but not denatured chains on the membrane. (3) Switch to a tagged bait detected with anti-tag antibody. (4) Run non-reducing gels (omit DTT/beta-mercaptoethanol) — the antibody stays as intact ~150 kDa IgG, well separated from most proteins.
Cross-Linking for Weak and Transient Interactions
Many biologically important protein-protein interactions are weak (Kd in the low micromolar to high nanomolar range) or transient — an E3 ligase touching its substrate for seconds, a kinase docking onto a scaffold. These complexes often dissociate during lysis and washing, producing false negatives.
Chemical cross-linking covalently locks interacting partners inside the intact cell before lysis. The cross-linker is added to live cells, permeates the membrane, and reacts with side chains in close proximity across a protein-protein interface. Once cross-linked, the complex survives harsh lysis.
Cross-Linker Selection Guide
| Cross-Linker | Spacer Arm | Reactive Toward | Cleavable? | Best Use Case |
|---|---|---|---|---|
| DSP (dithiobis(succinimidyl propionate)) | 1.2 nm | Primary amines (Lys, N-terminus) | Yes — DTT/TCEP reduces internal disulfide | The default choice for Co-IP-MS; cleavable, so peptides release as unmodified species [5] |
| DTME (dithiobismaleimidoethane) | 1.35 nm | Sulfhydryls (Cys) | Yes — DTT/TCEP | Complementary to DSP; targets Cys pairs at different distances |
| BS3 (bis-sulfosuccinimidyl suberate) | 1.14 nm | Primary amines | No | Non-cleavable control; membrane-impermeable version available for cell-surface interactions |
| Formaldehyde | ~0.2 nm (cross-links formed via methylene bridges) | Broad (Lys, Arg, Cys, His, Trp, N-terminus) | No (reversible only by boiling in SDS) | Chromatin IP (ChIP); very short-range, captures direct contacts |
A 2025 methodological advance — SureCLIP — pairs DSP cross-linking with DTT-mediated elution: live cells are DSP-treated, quenched, lysed in RIPA (now safe since complexes are locked), immunoprecipitated, and eluted with DTT, which cleaves the disulfide in DSP's spacer arm. The eluate contains only cross-linked prey proteins; antibody stays on the beads. The preprint reports hundreds of interactors per bait, including for nuclear envelope baits previously inaccessible to Co-IP.
Practical protocol: DSP stock in DMSO (50–100 mM), used at 0.5–2 mM on live cells in PBS for 30 minutes at room temperature, quenched with 20–50 mM Tris (pH 7.5). Over-cross-linking increases background and can mask epitopes — titrate the cross-linker concentration in a pilot experiment. Monoclonal antibodies are more susceptible to epitope masking than polyclonals. Researchers who prefer to avoid optimization overhead can use Creative Proteomics' crosslinking protein interaction analysis service, which includes cross-linker titration and LC-MS/MS identification.
From Co-IP to Mass Spectrometry
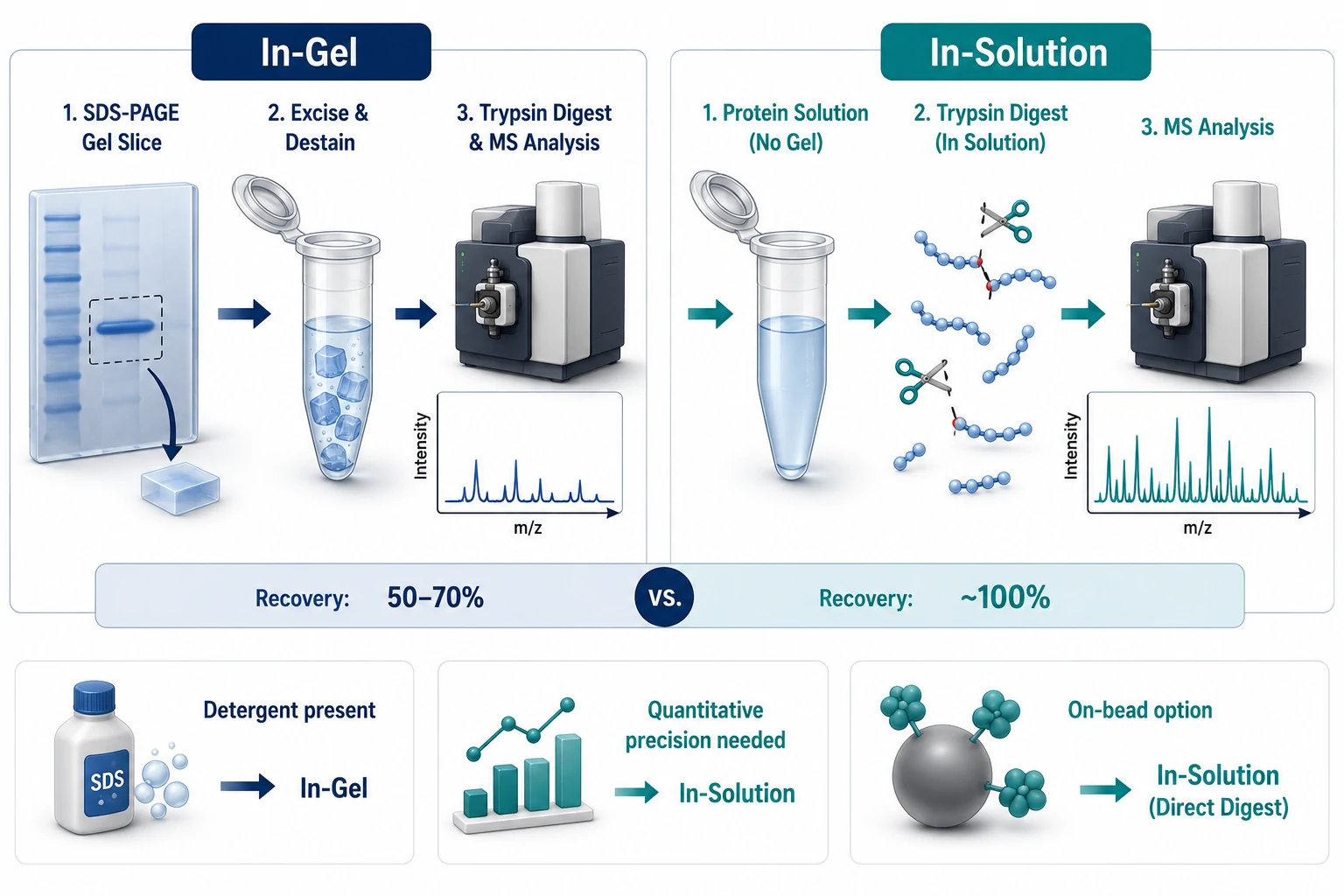
In-Gel vs In-Solution Digestion
Once the protein complex is captured and eluted, the sample must be proteolyzed into peptides for LC-MS/MS analysis. The decision between in-gel and in-solution trypsin digestion affects peptide recovery, contaminant carryover, and quantitative accuracy.
In-gel digestion resolves the eluate 1–2 cm into an SDS-PAGE gel, then the entire lane is excised, destained, reduced, alkylated, and digested with trypsin overnight. The gel serves as a built-in cleanup — detergents and salts wash away. The trade-off is peptide loss: recovery typically 50–70% [4], with hydrophobic peptides selectively lost. Creative Proteomics offers both in-gel and in-solution digestion services optimized for Co-IP samples, with protocols tailored to the detergent profile of each elution method.
In-solution digestion processes the eluate directly after reduction and alkylation. Recovery approaches 100%, but detergents (NP-40, SDS) suppress electrospray ionization and foul LC columns. Removal options include chloroform-methanol precipitation, SP3 paramagnetic bead cleanup, or MS-compatible detergents (RapiGest, ProteaseMAX). On-bead digestion — trypsin added directly to washed beads without elution — combines in-solution recovery with bead-based cleanup.
Decision rule: if the eluate is detergent-rich and protein is abundant, use in-gel. If quantitative precision across conditions is the goal, invest in detergent removal and use in-solution or on-bead digestion.
Quantitative Co-IP-MS Strategies
Distinguishing specific interactors from the "bead proteome" — proteins that bind non-specifically to beads, antibodies, or GST — is the central analytical challenge. Three quantitative strategies dominate:
Label-free quantification (LFQ) compares protein abundance between bait IP and control IP (IgG, GST-alone, or knockout) by spectral counting or XIC intensity. Hits typically require log2 fold change > 2 and adjusted p < 0.05 across triplicate experiments [2]. LFQ requires no metabolic labeling or expensive tags but needs more replicates for statistical power.
SILAC labels one cell population with heavy amino acids (13C6,15N2-Lys; 13C6,15N4-Arg) and another with light. Heavy and light IPs are mixed before digestion, and the H/L ratio directly quantifies enrichment. Because samples are mixed early, technical variation cancels, giving SILAC superior precision for distinguishing specific interactors from background [6]. Creative Proteomics offers SILAC-based proteomics analysis for quantitative Co-IP-MS experiments with built-in specificity controls.
TMT (tandem mass tags) multiplexes up to 16 conditions in one LC-MS/MS run via isobaric tags that release reporter ions during MS2 fragmentation. TMT excels at multi-condition comparisons — time courses, drug treatments, bait mutants — but reagent cost scales with sample number. For multi-condition interactome studies, TMT-based proteomics services enable multiplexed quantitative analysis with consistent batch-to-batch performance.
For Co-IP-MS discovery, Creative Proteomics provides integrated IP-MS protein interactomics solutions combining optimized capture with quantitative LC-MS/MS and interaction scoring.
Validation of Protein-Protein Interactions
A single Co-IP or pull-down band is a lead, not a conclusion. Three validation steps separate genuine interactors from artifacts:
Western blot confirmation comes first. Blot for the bait to confirm capture, then probe for the candidate interactor. Detection in the bait IP but not in the IgG or GST-alone control passes the first gate.
Reciprocal IP swaps bait and prey: if protein A pulls down B, IP B and blot for A. A reciprocal positive rules out antibody cross-reactivity and tag artifacts.
Functional validation asks whether the interaction matters biologically. Map the binding interface by deletion or alanine scanning, then show that interface mutations abolish both binding and downstream function — connecting physical interaction to mechanism. For comprehensive characterization, Creative Proteomics provides protein-protein interaction analysis services covering the full pipeline from capture to functional annotation.
Orthogonal biophysical methods — surface plasmon resonance (Biacore) or ITC — provide kinetic and thermodynamic parameters (kon, koff, Kd) that complement qualitative pull-down data. For deeper coverage of affinity purification-to-MS workflows, see our article on AP-MS for protein interaction mapping. When interacting partners occupy the same spatial neighborhood rather than binding directly, proximity labeling with BioID and TurboID provides an alternative that does not require the complex to survive lysis. Cross-linking strategies for stabilizing weak protein interactions share conceptual ground with RNA-protein cross-linking workflows that trap transient ribonucleoprotein assemblies.
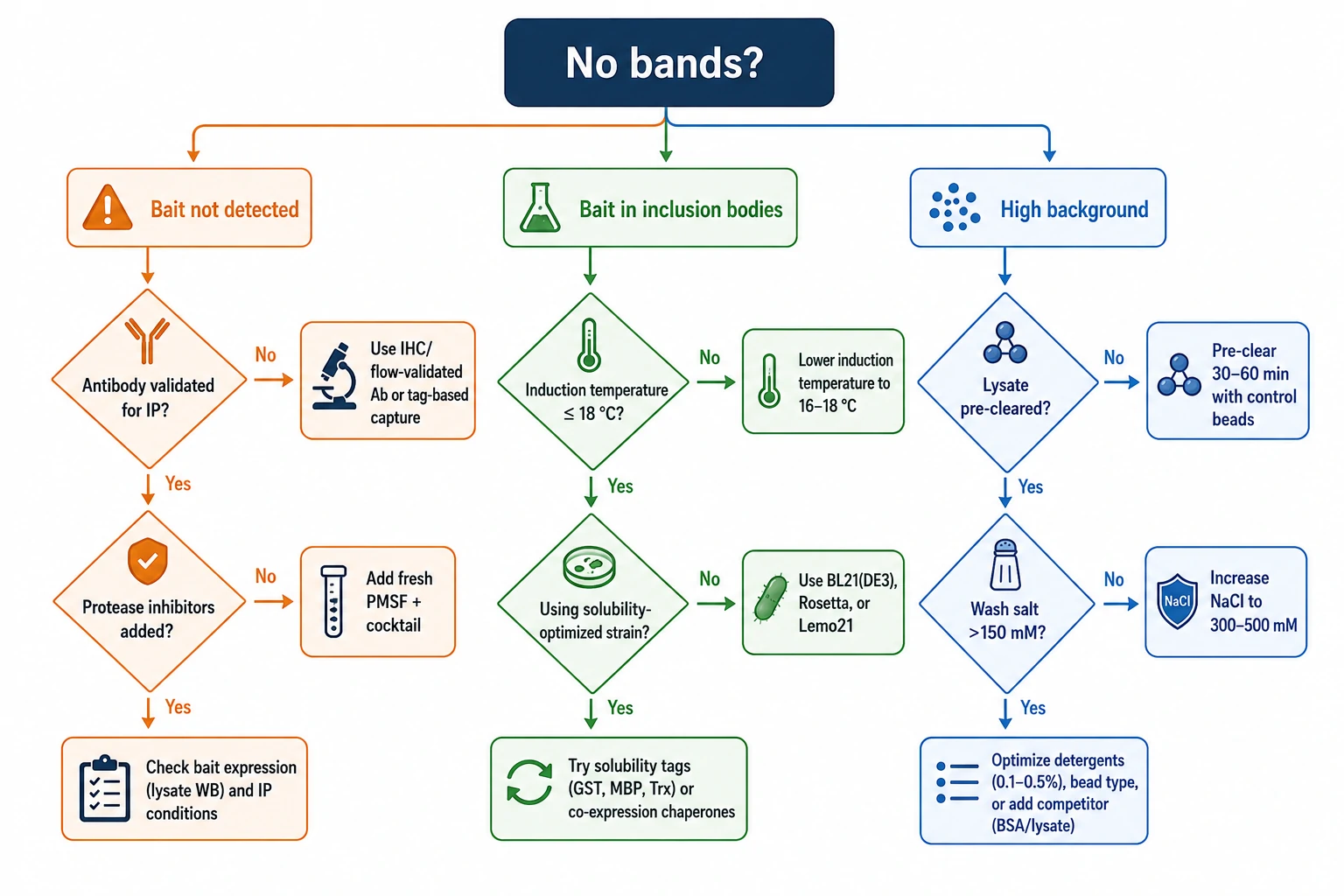
Troubleshooting Common Co-IP and GST Pull-Down Problems
| Problem | Root Cause | Solution |
|---|---|---|
| No bait detected after IP | Antibody does not recognize native conformation; bait degraded during lysis | Test antibody by native gel shift; add fresh protease inhibitors; pre-chill all buffers |
| No prey bands in pull-down | Interaction is weak/transient; bait misfolded | Add DSP cross-linking step before lysis; lower expression temperature for GST fusions; move tag to opposite terminus |
| Heavy chain (~50 kDa) masks bait | Antibody co-elutes and comigrates with target | Cross-link antibody to beads with DMP; use TrueBlot secondary; switch to tagged bait + anti-tag detection |
| High nonspecific background | Proteins binding beads or GST directly | Pre-clear lysate; increase wash salt to 300–500 mM; include GST-alone control; use magnetic beads |
| Interaction lost during wash | Wash conditions too stringent | Reduce salt; keep detergent at 0.1% or below; shorten wash time |
| GST bait in inclusion bodies | Expression temperature too high; codon bias | Induce at 18–25°C; reduce IPTG to 0.1 mM; test Rosetta or ArcticExpress strains; consider MBP or SUMO fusion |
| DnaK/GroEL contamination in GST pull-down | E. coli chaperones co-purify | Wash beads with ATP/Mg2+ buffer; use cleaner host strain |
| Low peptide recovery for MS | Detergent suppresses ionization; peptides lost in gel | Remove detergent by SP3 or precipitation before digestion; optimize on-bead digestion protocol; verify trypsin-to-protein ratio (~1:20 to 1:50) |
| Cross-linking causes epitope loss | Antibody binding site modified by cross-linker | Reduce cross-linker concentration; try polyclonal antibody; cross-link post-antibody-binding on beads |
FAQ
Q: What is the difference between Co-IP and GST pull-down?
A: Co-IP captures protein complexes from cell lysate using an antibody against the bait, preserving interactions that formed in the native cellular environment. GST pull-down uses a recombinant GST-fusion bait produced in E. coli to fish for interacting partners from a separately prepared lysate. Co-IP reflects in-cell interactions; GST pull-down identifies direct binding partners under controlled biochemical conditions.
Q: How much antibody should I use for a Co-IP?
A: Start with 1–5 ug of antibody per 500 ug to 1 mg of total lysate protein. Titrate downward — the minimum amount that captures the bait quantitatively minimizes heavy-chain co-elution and nonspecific background.
Q: How can I prevent antibody heavy and light chains from masking my target on Western blots?
A: Cross-link the antibody to Protein A/G beads with DMP or BS3 before the IP. The antibody stays on the beads during elution. Alternatively, use conformation-specific secondary antibodies (TrueBlot) or switch to a FLAG/HA-tagged bait detected with anti-tag antibodies.
Q: Which cross-linker is best for capturing weak protein interactions before Co-IP?
A: DSP (dithiobis(succinimidyl propionate)) is the default choice for Co-IP-MS. It has a 1.2 nm spacer arm, reacts with lysine primary amines, and is cleavable by DTT — the prey proteins release as unmodified species for downstream MS analysis.
Q: Can I use the same antibody for Western blot and Co-IP?
A: Not automatically. Many antibodies that work for Western blot recognize denatured epitopes exposed by SDS-PAGE. An antibody must be validated for IP or an application requiring native epitope recognition (IHC, flow cytometry) to work in Co-IP.
Q: How do I choose between in-gel and in-solution digestion for Co-IP-MS?
A: Use in-gel digestion when the eluate contains detergents that interfere with LC-MS and when protein amount is not limiting. Use in-solution or on-bead digestion when maximizing peptide recovery is the priority and detergents can be removed or avoided.
Q: What is the best tag for quantitative Co-IP-MS?
A: FLAG is the preferred single tag — 3×FLAG peptide elution produces the cleanest eluate without antibody contamination. FLAG-HA tandem tags provide even higher specificity through sequential purifications.
Q: How do I confirm my Co-IP worked before committing to an expensive MS run?
A: Run 10% of the IP eluate on a silver-stained or Coomassie-stained SDS-PAGE gel alongside the input lysate and the control IP. You should see enrichment of the bait band and additional bands in the bait IP lane that are absent from the control. If the bait and control lanes look identical, optimize capture before proceeding to MS.
*For research use only.
References:
- Rethi-Nagy Z, Abraham E, Lipinszki Z. GST-IVTT Pull-Down: Fast In Vitro Method for Validating and Mapping Protein–Protein Interactions. FEBS Open Bio, 2022, 12(11):1988-1995. doi:10.1002/2211-5463.13480 (CC BY 4.0)
- Fu Y, Li L, Zhang X, et al. Systematic HOIP interactome profiling across mouse tissues by affinity purification mass spectrometry. Nature Communications, 2024, 15:47289. doi:10.1038/s41467-024-47289-2 (CC BY 4.0)
- Sagrin MS, Tran H, Wilson MR, et al. In vivo Polycystin-1 interactome using a novel Pkd1 knock-in mouse model. PLOS ONE, 2023, 18(10):e0289778. doi:10.1371/journal.pone.0289778 (CC BY 4.0)
- Kaisar M, Armstrong J, van Rijswijk M, et al. Comparison of in-gel and in-solution proteolysis in the proteome profiling of organ perfusion solutions. Clinical Proteomics, 2023, 20:51. doi:10.1186/s12014-023-09440-x (CC BY 4.0)
- Shahinuzzaman M, Chowdhury SM, et al. Identification of Inflammatory Proteomics Networks of TLR4 through Immunoprecipitation-Based Chemical Cross-Linking Proteomics. Proteomes, 2022, 10(3):31. doi:10.3390/proteomes10030031 (CC BY 4.0)
- Wu S, Zhang S, Liu CM, Fernie AR, Yan S. Recent Advances in Mass Spectrometry-Based Protein Interactome Studies. Molecular & Cellular Proteomics, 2025, 24(1):100887. doi:10.1016/j.mcpro.2024.100887 (CC BY 4.0)













