RNA-Binding Proteins and Their Significance
RNA-binding proteins (RBPs) govern the lifecycle of every transcript — from splicing and nuclear export to cytoplasmic localization, translational control, and degradation. A single mRNA can be bound by dozens of RBPs, and the composition of that ribonucleoprotein (RNP) complex often determines whether the transcript is translated, stored, or degraded. RNA pull-down is the most direct experimental approach for identifying which proteins physically associate with a given RNA of interest.
RBPs constitute one of the largest protein classes in the human proteome, with over 1,500 experimentally validated members. They interact with RNA through a relatively small set of modular domains — RRM, KH domain, zinc finger, dsRBD, and DEAD-box helicase domains — but combinatorial diversity produces extraordinary functional specificity. The biological stakes of RBP dysregulation are high: mutations in FUS, TDP-43, and hnRNPA1 are implicated in the molecular pathology of amyotrophic lateral sclerosis through aberrant RNP aggregation; FMRP loss is associated with Fragile X syndrome, disrupting synaptic mRNA translation; and RBPs including LIN28, IGF2BP1-3, and HuR stabilize oncogenic transcripts in preclinical cancer models [1]. Identifying the full complement of proteins that bind a disease-associated RNA — particularly long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs) whose function often depends entirely on their protein partners — has become an essential experiment for functional characterization.
RNA Pull-Down Workflow Overview
The core RNA pull-down workflow comprises five stages, each with decision points that affect specificity, yield, and biological relevance:
1. Probe design and synthesis — Generate the RNA bait by in vitro transcription (IVT) or chemical synthesis, incorporating a label or affinity tag. 2. Protein extraction — Prepare cell or tissue lysate under conditions that preserve RNP integrity. 3. Cross-linking (optional but often essential) — Covalently trap weak or transient RNA-protein contacts. 4. Capture and washing — Immobilize the labeled RNA on an affinity matrix, incubate with lysate, and wash away non-specific binders. 5. Elution, detection, and identification — Release bound proteins for SDS-PAGE, Western blot, or LC-MS/MS analysis.
A critical but frequently overlooked step — negative controls — runs in parallel. Every RNA pull-down requires at least one negative control (antisense RNA, scrambled sequence, beads-only, or label-only) to distinguish specific interactors from background. Without proper controls, pull-down datasets are dominated by abundant non-specific RNA-binding proteins that obscure the specific interactome.
 Figure 1: RNA Pull-Down Pipeline Overview — Five-Stage Workflow Infographic
Figure 1: RNA Pull-Down Pipeline Overview — Five-Stage Workflow Infographic
RNA Probe Design and Preparation
IVT, Probe Length, and Structure
In vitro transcription (IVT) using T7, SP6, or T3 RNA polymerase is the standard method for generating RNA probes [3]. The DNA template — typically a PCR product with the polymerase promoter appended to the 5' end — is transcribed in the presence of NTPs and, for biotin-labeled probes, a fraction of biotin-UTP or biotin-CTP (10-30% of the corresponding unmodified NTP). Probe length is a practical variable: 200-500 nucleotides provides sufficient sequence context for secondary structure formation while remaining amplifiable by PCR. For long RNAs (>1 kb), tiling probes covering overlapping segments are preferable to a single full-length probe.
RNA structure matters — the probe must adopt a native or near-native fold to capture biologically relevant interactions. Standard practice includes a refolding step: heat-denature the RNA at 65-95 degrees C for 2-5 minutes in folding buffer (10-50 mM Tris, pH 7.4, 50-100 mM KCl, 1-5 mM MgCl2), then slow-cool to room temperature or 4 degrees C. For structured RNAs (ribozymes, IRES elements), magnesium concentration and cooling rate should be optimized empirically.
lncRNA and circRNA Considerations
lncRNAs present unique challenges: they are often low-abundance, >1 kb, and contain extensive secondary structure. Tiling probe sets — multiple overlapping biotinylated DNA oligonucleotides (20-25 nt each, spaced every 10-15 nt) that hybridize to the target — capture the endogenous lncRNA with its native RNP complex intact. This is the principle behind ChIRP and RAP-MS, both of which use tiling oligonucleotide probes rather than in vitro-transcribed RNA baits.
circRNAs require probes targeting the backsplice junction — the unique sequence created by the covalent ligation of the 5' and 3' ends during back-splicing. This distinguishes the circular isoform from the linear host gene transcript. Because circRNAs are resistant to exonucleases, pre-treatment with RNase R enriches for circular species prior to pull-down.
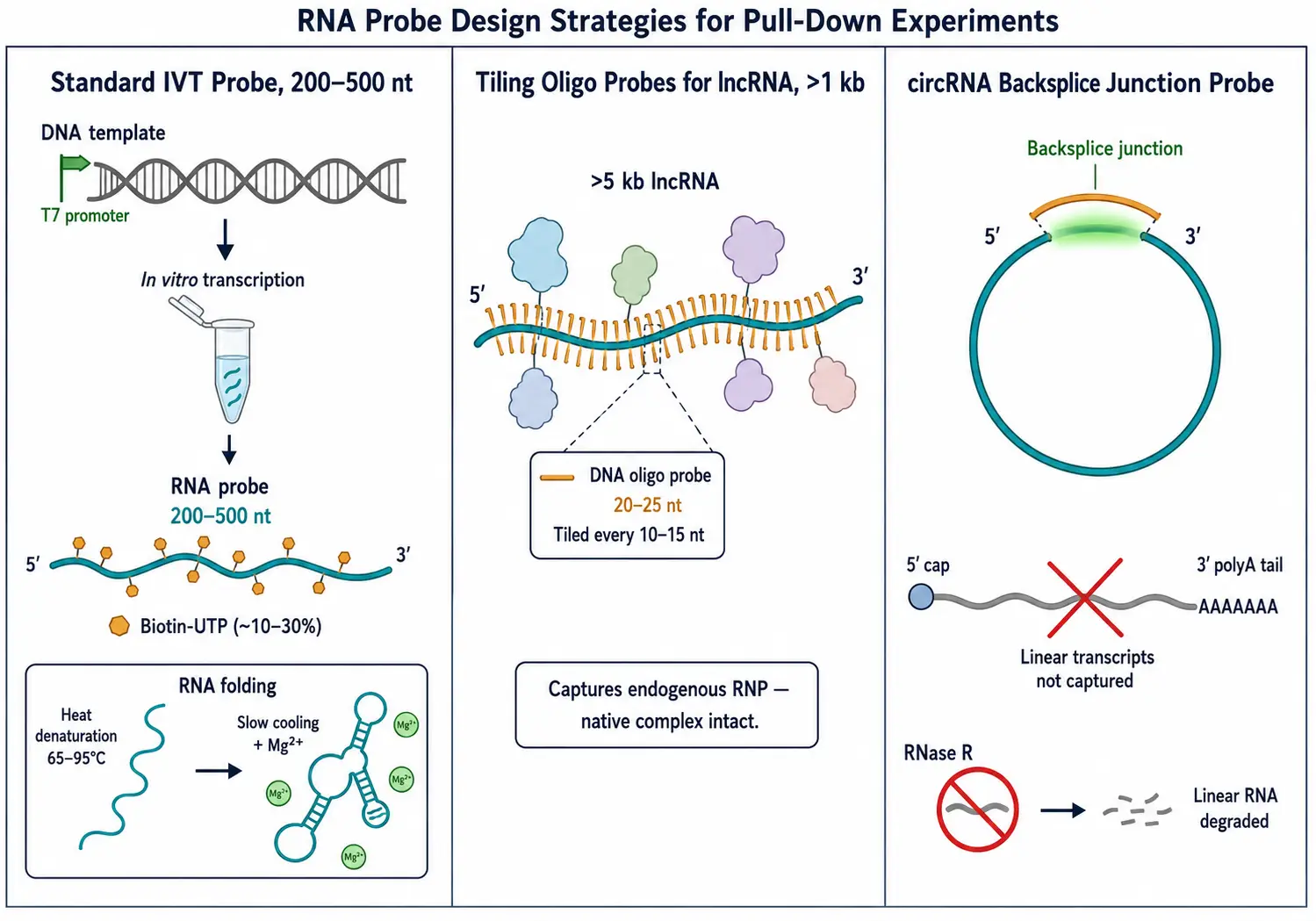 Figure 2: RNA Probe Design — IVT Synthesis, Biotin Incorporation, and Tiling Strategy for lncRNAs
Figure 2: RNA Probe Design — IVT Synthesis, Biotin Incorporation, and Tiling Strategy for lncRNAs
Labeling Strategy Comparison
Biotin, Digoxigenin, and Aptamer Tags
The choice of labeling strategy determines the affinity matrix, wash stringency, and background profile.
Biotin-streptavidin is the workhorse system. Biotinylated UTP or CTP is incorporated during IVT at a controlled ratio, and streptavidin-coated beads capture the biotinylated RNA with femtomolar affinity (Kd ~10^-15 M) — enabling stringent washes with up to 1 M NaCl and 0.5% detergent without dissociating the RNA. The trade-off is that biotin-streptavidin binding is essentially irreversible, requiring denaturing elution (SDS loading buffer at 95 degrees C).
Digoxigenin (DIG)-anti-DIG uses the hapten digoxigenin incorporated into the RNA and captured by anti-DIG antibodies. The affinity (Kd ~10^-9 to 10^-10 M) is weaker than biotin-streptavidin, permitting gentler elution under non-denaturing conditions — an advantage for recovering active protein complexes.
MS2 and PP7 aptamer tags exploit the high-affinity interaction between bacteriophage coat proteins and their cognate RNA stem-loops. The RNA of interest is expressed as a fusion with tandem MS2 stem-loops (6-24 copies), enabling in vivo RNP assembly prior to pull-down. The limitation is that the aptamer array may alter RNA folding or localization.
Tobramycin aptamer (J6f1) represents a minimalist alternative — a ~40-nt RNA aptamer with nanomolar affinity for tobramycin. The small footprint minimizes structural perturbation, making it attractive for highly structured RNAs. For step-by-step optimization of biotin-based pull-down conditions — including probe amount, buffer composition, and specificity verification — see the systematic comparison by Tsuji [5].
Selection Matrix
| Criterion | Biotin-Streptavidin | DIG-anti-DIG | MS2/PP7 Aptamer | Tobramycin Aptamer |
|---|---|---|---|---|
| Affinity | Femtomolar | Nanomolar | Nanomolar | Nanomolar |
| Wash stringency | Highest (1 M NaCl) | Moderate (0.3-0.5 M) | Moderate | Moderate |
| Elution | Denaturing only | Native or denaturing | Native (tag cleavage) | Native (competition) |
| Probe source | IVT only | IVT only | In vivo expression | IVT only |
| Aptamer footprint | N/A | N/A | Large (6-24x) | Small (~40 nt) |
| Best for | Discovery MS | Functional recovery | In vivo RNP capture | Structured RNAs |
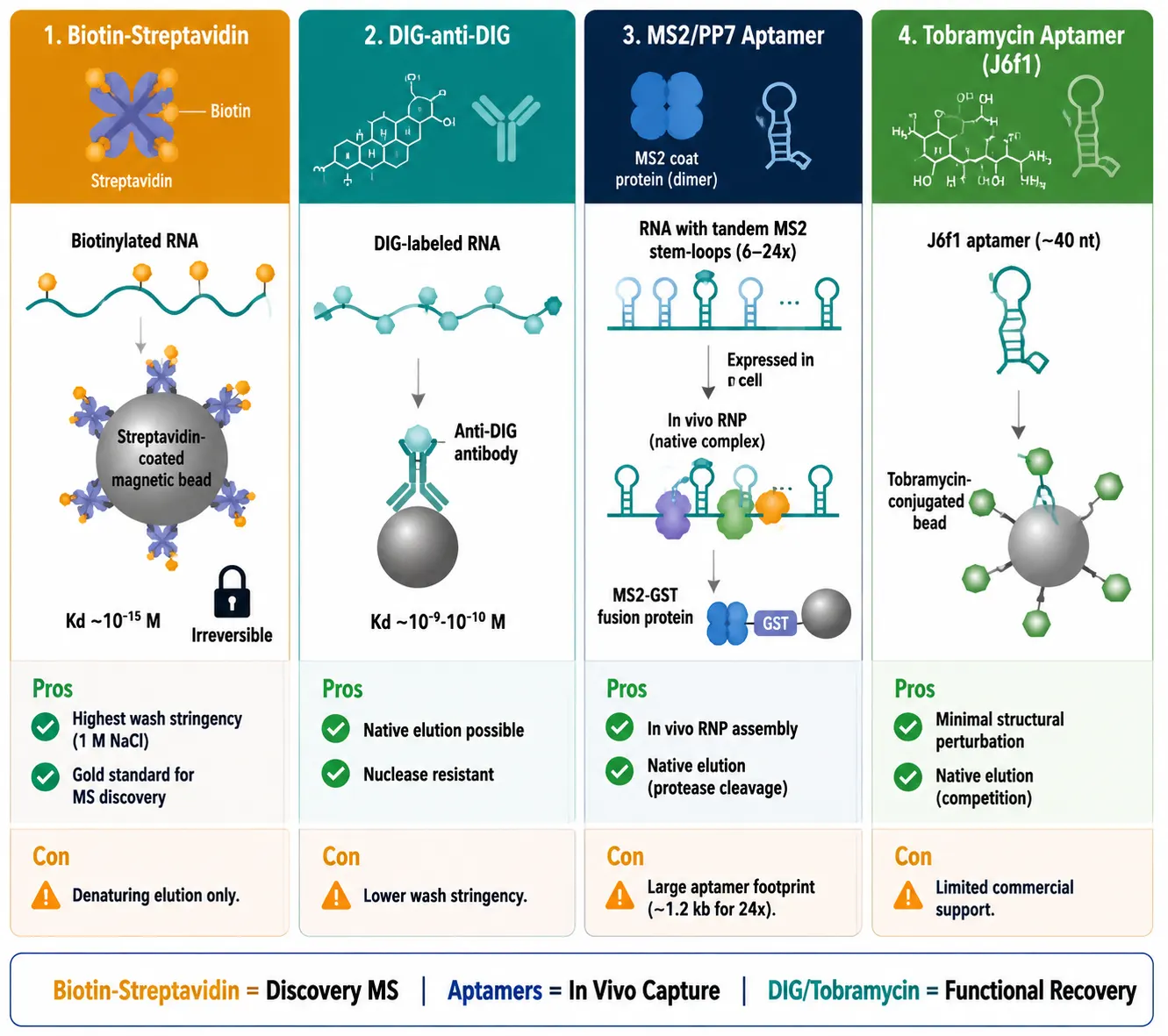 Figure 3: Labeling Strategy Selection Matrix — Biotin vs DIG vs MS2 vs Tobramycin
Figure 3: Labeling Strategy Selection Matrix — Biotin vs DIG vs MS2 vs Tobramycin
Protein Extraction and MS Preparation
The goal of protein extraction is to solubilize the proteome while preserving RNP integrity. Native lysis (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% NP-40, protease and RNase inhibitors) preserves protein complexes and RNA structure and is preferred for cytoplasmic RBPs. Denaturing lysis (8 M urea or 1% SDS followed by dilution to 0.1% SDS) releases proteins from all subcellular compartments but requires buffer exchange before incubation with the RNA bait.
For nuclear RBPs and chromatin-associated proteins, a two-step fractionation protocol substantially improves coverage. First, cytoplasmic proteins are extracted with a mild buffer (Buffer A: 10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.1% NP-40). The pelleted nuclei are then solubilized in a high-salt buffer (Buffer B: 20 mM HEPES, pH 7.9, 400 mM NaCl, 1 mM EDTA, 10% glycerol) to release chromatin-associated RNPs. The choice between RIPA buffer (high SDS, denaturing) and mild NP-40 buffer (native) directly determines which RNP subpopulations are recovered: RIPA releases chromatin-bound RBPs but strips weakly associated factors; NP-40 preserves weak interactions but underrepresents the nuclear RBP pool. When the subcellular localization of the target RNA is unknown, running parallel native and denaturing extractions from the same sample provides the most complete RBP coverage.
Pre-clearing the lysate with empty streptavidin beads for 30-60 minutes before incubation removes endogenous biotinylated proteins and bead-binding proteins — a simple step that substantially reduces background. For quantitative MS workflows, our quantitative proteomics platform supports SILAC, TMT, and label-free approaches for comparing pull-down conditions.
Pull-Down: Incubation to Elution
Bead Selection
Magnetic beads (1-5 μm, streptavidin-coated) offer rapid magnetic collection, minimal non-specific binding, and excellent reproducibility — the preferred format for quantitative MS. Agarose resin (50-150 μm) provides higher binding capacity and is preferred for large-scale preparative pull-downs but traps more non-specific proteins. Streptavidin-coated magnetic beads are the default for analytical-scale experiments, with 10-50 μL of bead slurry per pull-down and bead pre-blocking in 0.1% BSA or 0.02% tRNA. For a broader discussion of bead-based affinity purification in protein-centric workflows — including magnetic vs. agarose trade-offs and wash stringency optimization — see our guide on AP-MS for protein interaction mapping.
Cross-Linking for Weak Interactions
Weak or transient RNA-protein interactions require covalent stabilization. UV cross-linking (254 nm) induces zero-length covalent bonds at direct contact points, capturing only direct RNA-protein contacts with high specificity. Efficiency varies with nucleotide and amino acid composition — pyrimidines (uracil) and aromatic residues cross-link most efficiently. Formaldehyde cross-linking (0.1-0.2%, 10-30 min) captures both direct and indirect interactions, preserving the entire RNP complex. Formaldehyde penetrates tissues more effectively, making it preferred for tissue samples. A practical framework: use UV for direct binders, formaldehyde for full complex characterization, and both in parallel for maximum coverage. For protein-protein cross-linking strategies applicable to RNP complex stabilization, refer to our resource on cross-linking mass spectrometry for interactomics.
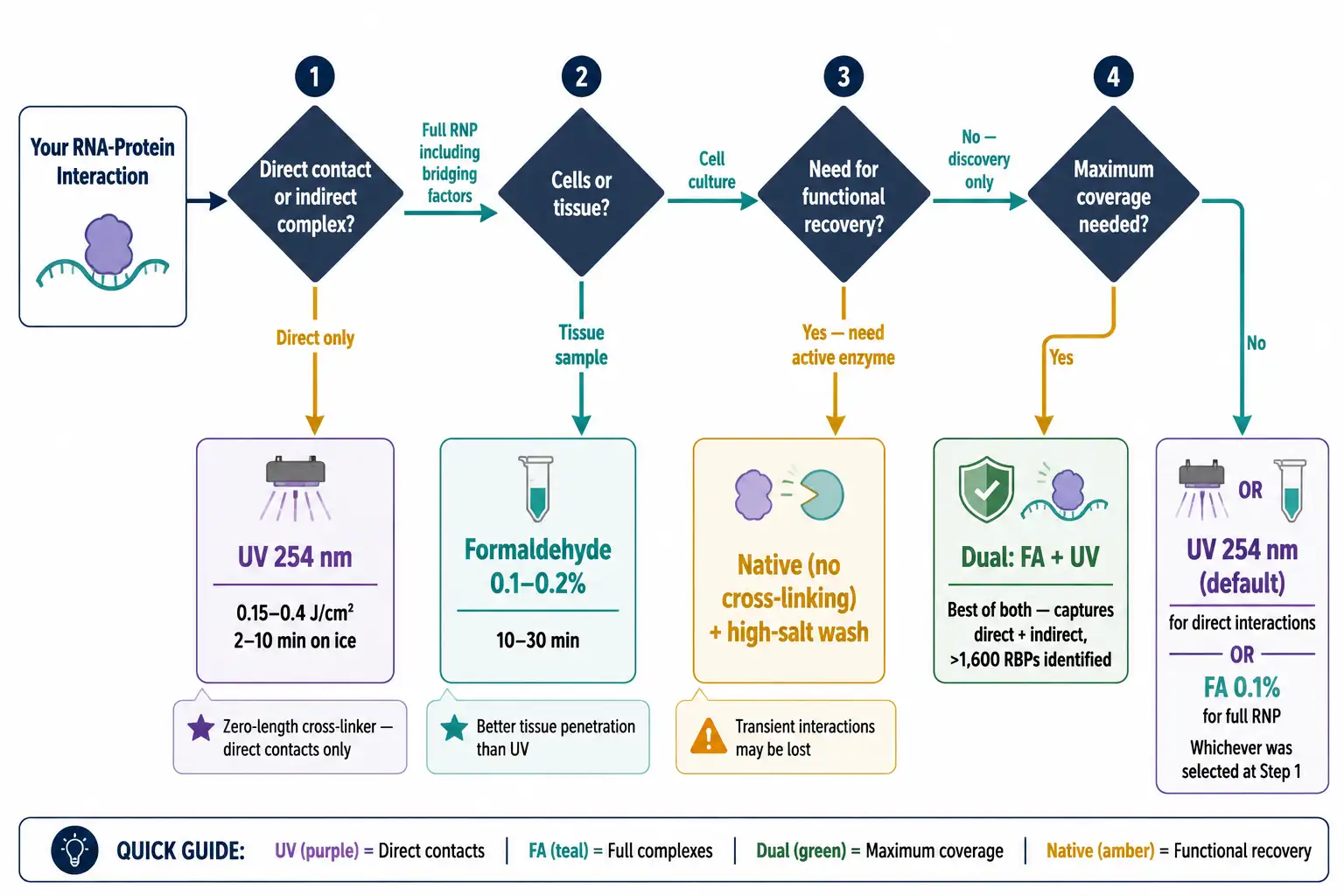 Figure 4: Cross-Linking Strategy Decision Tree — UV vs Formaldehyde vs Dual
Figure 4: Cross-Linking Strategy Decision Tree — UV vs Formaldehyde vs Dual
Elution Methods
For SDS-PAGE and Western blot, boil beads in 1x SDS-PAGE loading buffer at 95 degrees C for 5-10 minutes. For MS, on-bead digestion is preferred: after the final wash, proteins are reduced (DTT/TCEP), alkylated (iodoacetamide), and digested with trypsin directly on the beads. The resulting peptides are desalted and analyzed by LC-MS/MS — analyzed via our protein identification service with DDA or DIA acquisition. For functional recovery, competitive elution with excess free biotin (10-50 mM) can displace biotinylated RNA-protein complexes, though efficiency is modest due to the slow off-rate.
Negative Controls
A pull-down without proper negative controls is uninterpretable. At minimum, include: Antisense or scrambled RNA — identical length and nucleotide composition but lacking the specific sequence or structural motifs of the target RNA. Beads-only control — streptavidin beads incubated with lysate without any RNA probe, identifying proteins that bind to the bead surface or streptavidin. Label-only control — beads incubated with free biotin or an irrelevant biotinylated oligonucleotide. Label-swap control — an unrelated RNA probe with the same label to confirm sequence-specific enrichment.
In quantitative MS workflows (SILAC, TMT), these controls are incorporated into the experimental design, and specific interactors are identified by their enrichment ratio relative to the control channel. A fold-change threshold of ≥2 with FDR ≤0.05 is a common starting point, but the appropriate threshold depends on the signal-to-noise characteristics of the specific pull-down.
RBP Identification by Mass Spectrometry
LC-MS/MS is the standard platform for unbiased identification of proteins from RNA pull-down eluates. After on-bead trypsin digestion, peptides are separated by reversed-phase nano-LC and analyzed by DDA or DIA on a high-resolution mass spectrometer (Q-TOF or Orbitrap). For comparative pull-down experiments (target RNA vs. control), three quantitative approaches reduce the false discovery rate:
SILAC — Cells are grown in "light" (Arg0/Lys0) or "heavy" (Arg10/Lys8) medium, and labeled lysates are mixed prior to pull-down. The heavy:light ratio directly reports enrichment of each protein. TMT — Proteins are digested, labeled with isobaric tags, pooled, and analyzed simultaneously, enabling up to 18-plex multiplexing in a single MS run. Label-free quantification (LFQ) compares peptide peak areas across separate runs — simpler but lower precision. Quantitative MS strategies for interactomics — including SAINT scoring, CRAPome filtering, and statistical frameworks for distinguishing specific from non-specific interactions — are discussed in our AP-MS resource. Our SILAC and TMT quantification services support all three approaches with statistical filtering and functional annotation.
Regardless of the quantification strategy, the data analysis pipeline should include: subtraction of proteins identified in the negative control, fold-change filtering (≥2-3 fold), statistical testing (moderated t-test or empirical Bayes), and functional annotation (GO enrichment, protein interaction network analysis). For a comprehensive review of MS platforms and workflows for RBP detection, see Moradi et al. [2].
Validation — Confirming MS Findings
MS identification from a pull-down experiment is a discovery result, not a conclusion. Every candidate interactor requires orthogonal validation.
Western blot is the most direct validation. Load 20-30% of the pull-down eluate per lane alongside 5% input lysate as loading control; probe for the candidate RBP; strip and re-probe for a known non-RNA-binding protein (e.g., GAPDH, tubulin) as a negative validation control. A specific band in the target RNA lane that is absent or substantially weaker in the scrambled RNA and beads-only lanes constitutes positive validation. For quantitative assessment, band intensity can be normalized to input and expressed as fold-enrichment over the scrambled control.
RIP-qPCR is the reciprocal experiment: immunoprecipitate the candidate protein and ask "does the target RNA co-immunoprecipitate?" A fold enrichment ≥3 over an IgG isotype control with Ct values<30 in the target IP is a reasonable positivity threshold. Include a negative control RNA (e.g., GAPDH mRNA for a non-ribosomal candidate) to confirm the interaction is specific rather than reflecting general RNA-binding activity.
RNA EMSA confirms direct binding with purified components. Incubate recombinant candidate RBP (typically 0.1-5 μM) with labeled RNA probe (1-10 nM) in binding buffer (10-20 mM Tris, pH 7.5, 50-100 mM KCl, 1-5 mM MgCl2, 0.01-0.1 mg/mL BSA, 2-5% glycerol), resolve on a 4-6% native polyacrylamide gel, and detect the shifted complex. For a biologically meaningful interaction, the apparent Kd should fall in the nanomolar to low micromolar range; binding weaker than ~10 μM is likely non-specific. Competition with 50-100x excess unlabeled wild-type RNA (shift disappears) versus mutant RNA with the predicted binding motif disrupted (shift persists) demonstrates sequence-specificity. For proteins identified through pull-down assay screening, these three validation approaches — Western blot for specificity, RIP-qPCR for reciprocity, and EMSA for direct binding — form the standard confirmation package.
In Vitro vs. In Vivo RNA Pull-Down
In vitro RNA pull-down uses an exogenously synthesized RNA probe incubated with lysate. It is the most accessible format — no genetic manipulation required, probe synthesis within days, and conditions fully controllable. The limitation is that the RNA may not fold correctly in lysate, and proteins that only associate with the RNA in the context of intact cellular architecture may be missed.
In vivo RNA pull-down captures endogenous RNPs assembled in living cells, either through expression of aptamer-tagged RNA (MS2, PP7, tobramycin) or hybridization-based methods (ChIRP, RAP-MS) that use tiling antisense oligonucleotides to capture endogenous, unmodified RNA from cross-linked cell lysates. In vivo approaches preserve native RNP stoichiometry but require more complex experimental setups. For most discovery applications, an in vitro pull-down followed by in vivo validation of key candidates provides the best balance of throughput and biological relevance.
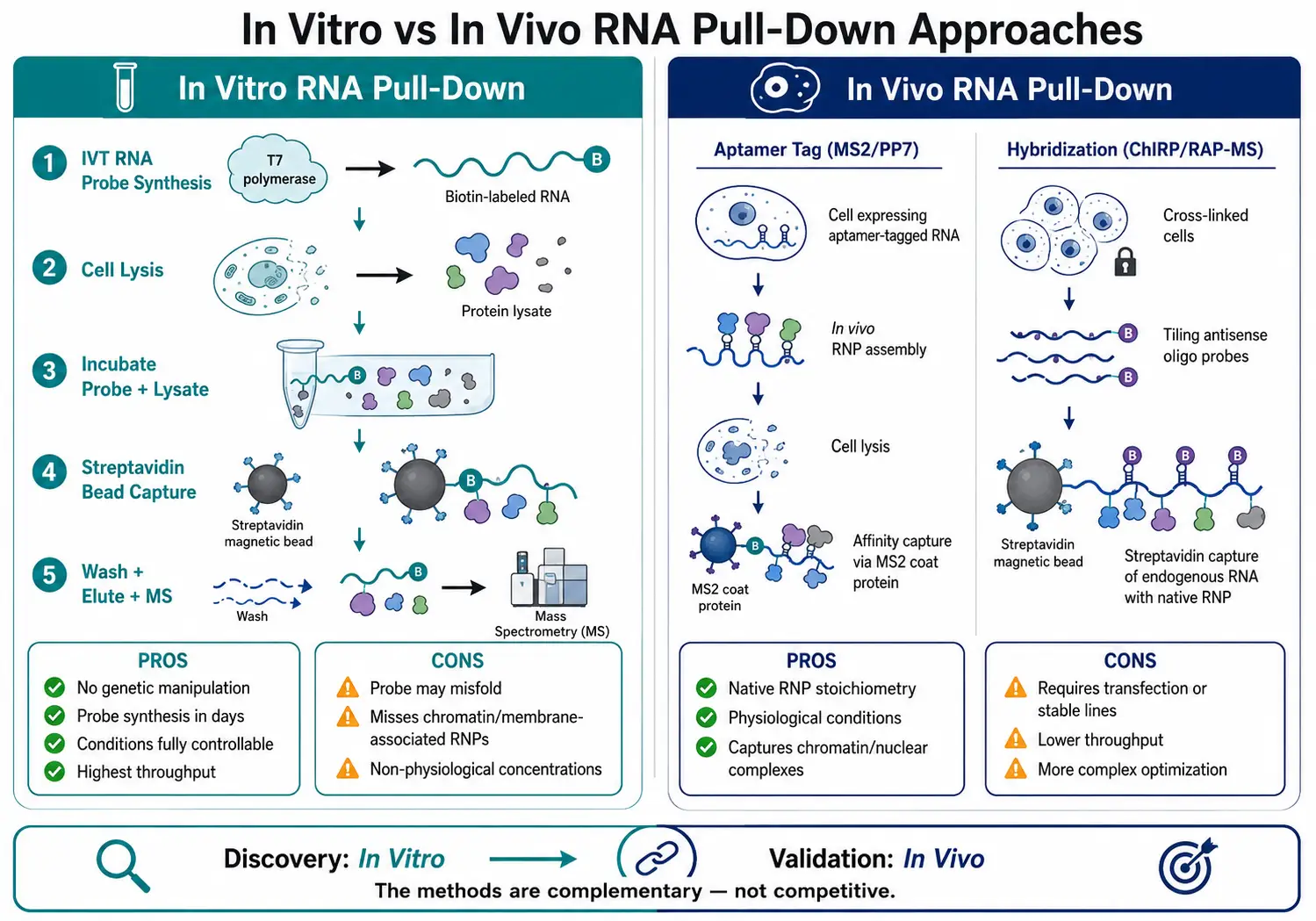 Figure 5: In Vitro vs. In Vivo RNA Pull-Down Comparison — Workflow and Decision Framework
Figure 5: In Vitro vs. In Vivo RNA Pull-Down Comparison — Workflow and Decision Framework
RNA Pull-Down vs. RIP vs. CLIP-Seq
These three methods address different questions, and confusing them is a common source of experimental design errors.
| Feature | RNA Pull-Down | RIP | CLIP-Seq |
|---|---|---|---|
| Starting point | RNA of interest | Protein of interest | Protein of interest |
| Question asked | "Which proteins bind this RNA?" | "Which RNAs bind this protein?" | "Where on the RNA does this protein bind?" |
| Cross-linking | Optional (UV or FA) | Optional (native) or FA | UV (254 nm) — mandatory |
| Resolution | Protein-level | Transcript-level | Nucleotide-level |
| Throughput | Low (1 RNA per experiment) | Medium (transcriptome-wide) | High (binding site maps) |
| Best for | Characterizing novel ncRNAs | Profiling known RBP targets | Defining RBP binding motifs |
RNA pull-down is the method of choice when the research question is RNA-driven — for example, "What proteins bind to this lncRNA upregulated in metastatic tumors?" RIP and CLIP-seq are appropriate when the question is protein-driven. In practice, the methods are complementary: RNA pull-down identifies candidate RBPs for a newly discovered transcript, and CLIP-seq maps the binding sites of the most interesting candidates across the transcriptome. For a detailed comparison of RNA-centric and protein-centric interactome capture approaches, see the 2025 review by Yi and Yan [1] and the viral RNA interactome guide by Hanson et al. [4].
Troubleshooting Common RNA Pull-Down Problems
| Problem | Probable Cause | Solution |
|---|---|---|
| High background in control lane | Insufficient washing; bead non-specific binding | Increase wash salt to 0.5-1 M NaCl; pre-clear lysate with empty beads; add competitor tRNA (0.1 mg/mL) to binding buffer |
| No specific bands detected | RNA probe degraded; RBP below detection limit | Verify RNA integrity on denaturing gel; concentrate lysate or increase scale; add co-factors (ATP for helicases, heme for IRP) |
| RNA degradation during incubation | RNase contamination | Use RNase-free reagents; add RNasin or vanadyl ribonucleoside complexes; work at 4 degrees C |
| Probe does not bind beads | Insufficient biotin incorporation | Optimize biotin-UTP:UTP ratio (10-30%); pre-wash beads to remove preservatives |
| MS only identifies ribosomal proteins | Specific interactors masked by background | Use quantitative MS (SILAC/TMT); add heparin in washes; apply fold-change filtering with FDR control |
| Interaction validated by MS but not WB | Protein below WB detection limit; antibody epitope masked | Scale up pull-down 2-5x; use different antibody; include protease inhibitors throughout |
| RNA structure blocks protein binding | Probe misfolded; incorrect buffer | Optimize refolding protocol (temperature, Mg2+, cooling rate); use smaller RNA fragments |
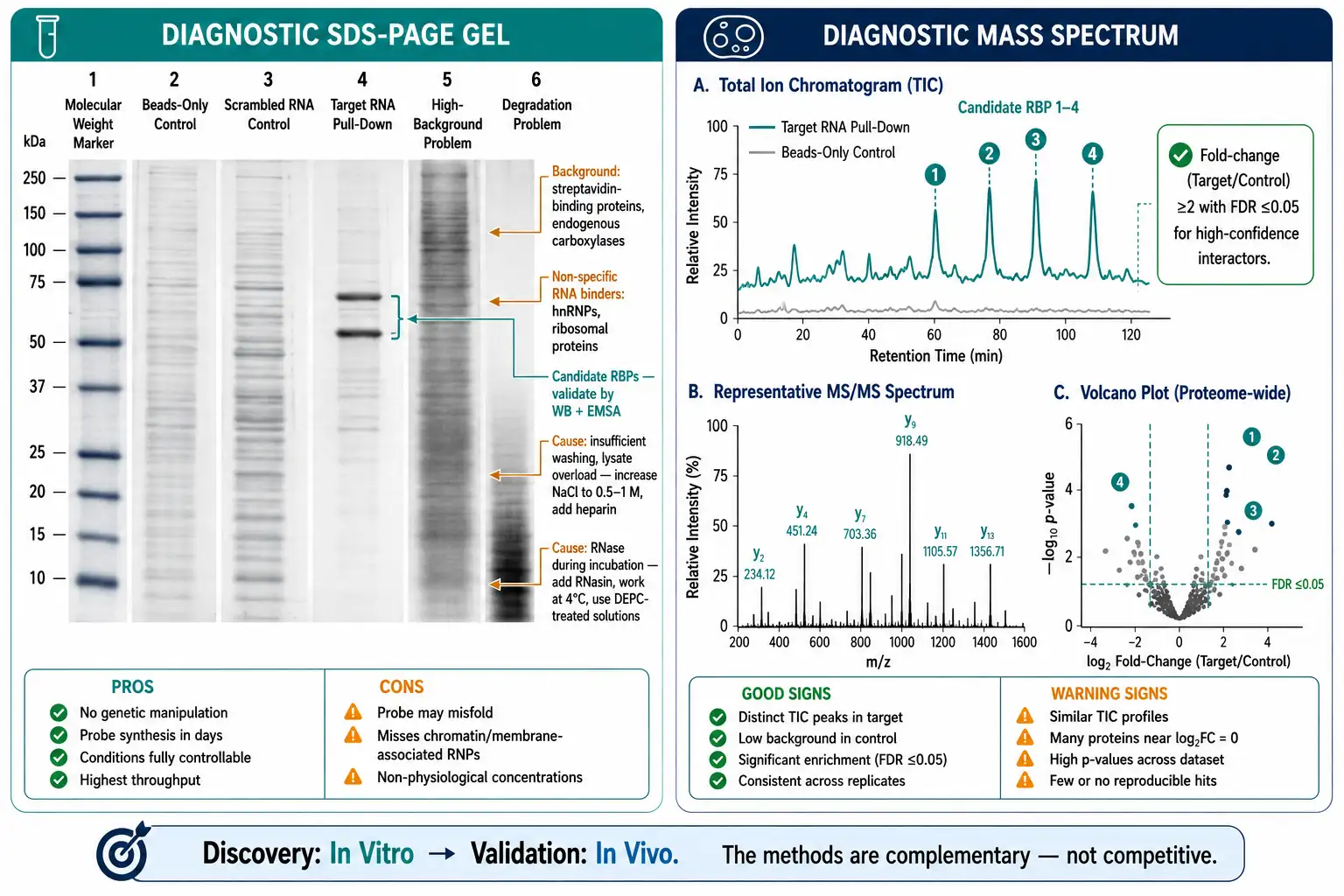 Figure 6: Annotated RNA Pull-Down Troubleshooting Guide — Diagnostic Gel and MS Spectrum
Figure 6: Annotated RNA Pull-Down Troubleshooting Guide — Diagnostic Gel and MS Spectrum
FAQ
How do I choose between biotin and MS2 labeling for my RNA pull-down?
Choose biotin-streptavidin when you need the simplest, most robust workflow for in vitro pull-down from lysate — it is the best-validated system with the highest affinity. Choose MS2 (or PP7) aptamers when you need the RNA-protein complex to assemble in living cells prior to capture, or when you want to recover native complexes by gentle elution.
Do I need to cross-link? When should I?
Cross-link when your interaction is weak or transient (off-rate faster than the pull-down incubation time), when the RNA is highly structured and the protein binds a specific conformation, or when working with tissue samples. Skip cross-linking when validating a known strong interaction by Western blot or when you need enzymatically active proteins for functional assays.
How many biological replicates do I need for MS-based RNA pull-down?
For label-free quantification: minimum three biological replicates per condition (target RNA plus at least one negative control). For SILAC or TMT-based quantification: two to three replicates, as the internal standard reduces technical variability.
My RNA pull-down worked by Western blot but MS only identified abundant non-specific proteins. What went wrong?
Your specific interactor is likely low-abundance and buried under a background of ribosomal proteins and hnRNPs. Switch to quantitative MS (SILAC or TMT) to filter by enrichment ratio, increase wash stringency with heparin or high salt, pre-clear lysate with empty beads plus competitor tRNA at 0.1 mg/mL, and pool multiple pull-down replicates.
Can I pull down a circular RNA?
Yes. Design the probe to span the backsplice junction — this ensures capture of the circular isoform and not the linear host transcript. Pre-treatment of total RNA with RNase R enriches for circular species and improves pull-down specificity.
My lncRNA is longer than 5 kb. Should I make one long probe or multiple shorter ones?
Multiple shorter tiling probes (20-25 nt DNA oligonucleotides spaced every 10-15 nt). A single 5 kb IVT RNA probe will almost certainly misfold. Tiling probes hybridize to the endogenous lncRNA, capturing it with its native RNP complex intact — the approach used by ChIRP and RAP-MS.
How do I distinguish direct from indirect RNA-binding proteins in my pull-down?
UV cross-linking (254 nm) captures only direct RNA-protein contacts — it is a zero-length cross-linker. If a protein appears in a UV-cross-linked pull-down, it was in direct contact with the RNA. If it appears only in a formaldehyde-cross-linked pull-down, it may be indirectly associated. For the strongest evidence of direct binding, validate with recombinant protein by RNA EMSA.
References:
- Yi W, Yan J. Decoding RNA-protein interactions: methodological advances and emerging challenges. Advanced Genetics. 2025;6(2):2500011. (CC BY 4.0)
- Moradi M, Farjami Z, Akbarin MM. Spectrometry and its application for the detection of RNA-binding proteins: advancements, techniques and challenges. Analytical Science Advances. 2025;6(2):e70026. (CC BY 4.0)
- Jiang L, Yang J, He R, Zhu Y, Wang D. Protocol for detecting lncRNA-protein interactions in vitro by tRSA RNA pull-down assay. STAR Protocols. 2024;5(1):102818. (CC BY 4.0)
- Hanson WA, Romero Agosto GA, Rouskin S. Viral RNA interactome: the ultimate researcher's guide to RNA-protein interactions. Viruses. 2024;16(11):1702. (CC BY 4.0)
- Tsuji Y. Optimization of biotinylated RNA or DNA pull-down assays for detection of binding proteins: examples of IRP1, IRP2, HuR, AUF1, and Nrf2. International Journal of Molecular Sciences. 2023;24(4):3604. (CC BY 4.0)









