Beyond Ser/Thr/Tyr
If you have run a standard phosphoproteomics experiment — TiO₂ or IMAC enrichment at low pH, DDA or DIA acquisition on a Q-Exactive or timsTOF, MaxQuant or Spectronaut search against the human proteome with variable phosphorylation on S/T/Y — you have almost certainly missed something. Something large.
A 2024 study using strong anion exchange-based enrichment at neutral pH revealed that non-canonical phosphosites — phosphorylation on histidine, cysteine, aspartate, glutamate, lysine, and arginine — number approximately one-third as many as canonical pSer/pThr/pTyr sites under basal conditions. One-third. That figure rewrites the estimated size of the functional phosphoproteome. It means that for every three regulatory phosphorylation events you have mapped, there is roughly one more that standard workflows have systematically destroyed or ignored.
This is not an incremental refinement of phosphoproteomics. It is a blind spot the size of a subfield. The analytical problem has a simple chemical root: the P–N bonds in phosphohistidine (pHis), phospholysine (pLys), and phosphoarginine (pArg), the P–S bond in phosphocysteine (pCys), and the acyl-phosphate bonds in phosphoaspartate (pAsp) and phosphoglutamate (pGlu) are all acid-labile. The 0.1% trifluoroacetic acid or 0.1% formic acid used in every step of a standard phosphoproteomics workflow — from peptide desalting to IMAC loading to LC mobile phases — hydrolyzes these bonds within minutes to hours. Standard enrichment with TiO₂ or IMAC at pH 2.5–3.0 is a chemical selection against the very modifications you might most want to find.
The biology, meanwhile, has moved far ahead of the analytical chemistry. Histidine phosphorylation drives bacterial two-component signaling, the dominant signal transduction system in prokaryotes, and in mammals the NME family kinases transfer phosphate to histidine residues on substrates including the potassium channel KCa3.1, histone H4, and the GTPase Dynamin-1. Pyrophosphorylation of serine — the addition of a diphosphate group to generate ppSer — has been identified on over 700 sites in human cells, primarily on acidic serine-rich motifs. Phosphocysteine appears in the active sites of protein tyrosine phosphatases as a catalytic intermediate. These are not exotic modifications found in niche organisms; they occur in human cells, under physiological conditions, and evidence for their regulatory roles is accumulating rapidly.
This article provides a practical guide to the chemistry, enrichment, detection, and analysis of non-canonical phosphorylation. The focus is on what a research group evaluating their options needs to know: which modifications matter, why standard methods fail, what alternatives exist, and what the data can and cannot tell you. For groups running or outsourcing phosphoproteomics, Creative Proteomics provides phosphoproteomics services with method development capabilities that extend beyond canonical workflows.
Chemistry of Non-Canonical Phosphorylation
The chemical bond connecting phosphate to an amino acid side chain determines everything: stability, detection strategy, enrichment compatibility, and biological interpretation. The twenty proteinogenic amino acids present nine potential phosphorylation targets, but the chemistry divides them into four fundamentally distinct classes.
Phosphoramidate: pHis, pLys, pArg
The N-phosphate linkage — a phosphoramidate bond in which phosphorus bonds to a side-chain nitrogen atom — is the most biologically prominent class of non-canonical phosphorylation and the most analytically challenging.

Histidine phosphorylation produces two thermodynamically distinct isomers: 1-phosphohistidine (π-pHis, phosphorylation at the N1 position of the imidazole ring) and 3-phosphohistidine (τ-pHis, at the N3 position). Under physiological pH, 3-phosphohistidine is more stable than 1-phosphohistidine, and both are substantially more labile than phosphoserine. The phosphoramidate P–N bond has a standard free energy of hydrolysis (ΔG°') of approximately −12 to −14 kcal/mol, compared to approximately −6.5 to −9 kcal/mol for phosphoserine's P–O ester bond. This high-energy character is precisely what makes pHis an effective phosphoryl donor in bacterial phosphorelay systems, and precisely what makes it vanish under standard acidic conditions.
Phospholysine and phosphoarginine share the phosphoramidate chemistry but differ in biological abundance. pLys has been detected on histones and in nucleotide-binding proteins. An antibody-based enrichment method using anti-pHis monoclonal antibodies captures pHis-modified peptides but does not cross-react with pLys or pArg, leaving these residues largely inaccessible outside of SAX-based methods.
Acyl Phosphate: pAsp and pGlu
Phosphoaspartate and phosphoglutamate form through a mixed anhydride linkage — phosphate esterified to the carboxyl side chain. This acyl-phosphate bond is even more acid-labile than the phosphoramidate. Half-lives at pH 3.0 and 25°C can be under 30 minutes for pAsp in unstructured peptides, though local protein context modulates stability. pAsp is the central phospho-species in bacterial chemotaxis (the CheA-CheY two-component system) and in the catalytic cycle of P-type ATPases including the Na⁺/K⁺-ATPase and Ca²⁺-ATPase. pGlu appears in several prokaryotic signaling systems and has been detected in mammalian cell extracts, though its biological prevalence remains less well characterized than pHis.
Phosphorothioate: pCys
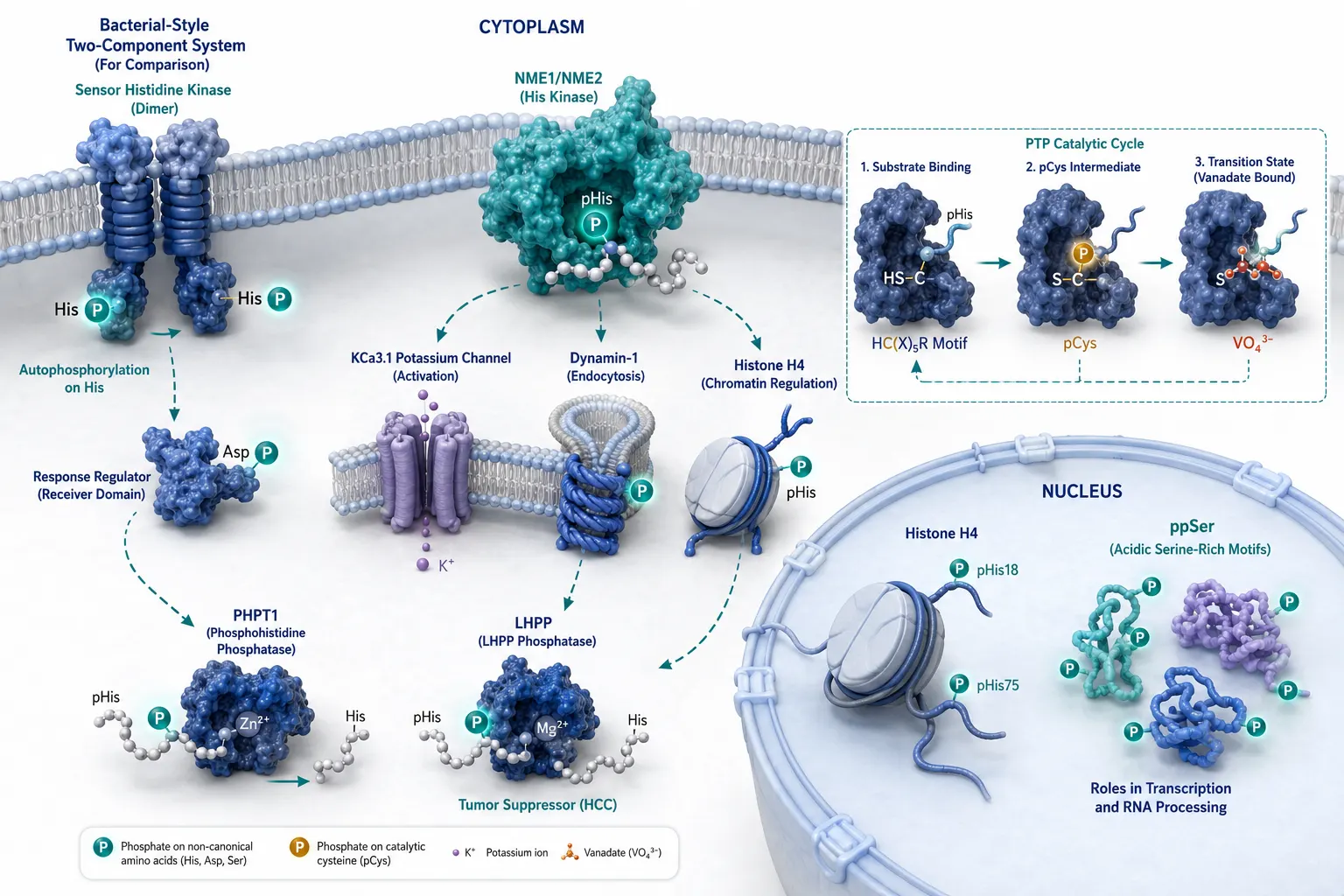
The P–S bond of phosphocysteine is chemically distinct from both N- and O-phosphate linkages. Its acid lability falls between that of phosphoramidates and acyl phosphates — measurable half-lives at pH 3.0 in the range of hours for some sequence contexts. pCys appears in the conserved HC(X)₅R catalytic motif of protein tyrosine phosphatases (PTPs), where it serves as a transient covalent intermediate during catalysis. Detection is complicated by the fact that the catalytic intermediate is intentionally short-lived; trapping strategies using vanadate or tungstate as phosphate analogs stabilize the pCys intermediate for analysis. Beyond PTP active sites, evidence for regulatory pCys in other contexts is emerging but sparse — though this may partly reflect detection difficulty rather than biological rarity.
Pyrophosphorylation: ppSer
Pyrophosphorylation attaches a diphosphate group to a serine hydroxyl, creating a phosphoanhydride-linked modification. First characterized comprehensively in a 2019 study reporting over 700 unique ppSer sites in human cells, ppSer is enriched on acidic serine-rich motifs and appears to be installed non-enzymatically under conditions of high intracellular diphosphate concentration. The P–O–P linkage has distinct stability properties: more acid-labile than pSer monoester but more stable than phosphoramidates under certain pH conditions. The functional significance of ppSer remains under active investigation; its enrichment on proteins involved in transcription, RNA processing, and ribosome biogenesis suggests roles in nuclear processes.
Acid Lability and Analytical Consequences
The practical implication of these chemistries is stark. The workflow that has generated the vast majority of the published phosphoproteome — protein extraction in denaturing buffers, reduction and alkylation, trypsin digestion, C18 desalting in 0.1% TFA, TiO₂ or IMAC enrichment in 0.1% TFA or 0.1% formic acid, LC separation in 0.1% formic acid — exposes peptides to acidic pH at every step from lysis to ionization.
For pSer, pThr, and pTyr, this is unproblematic. For pHis, pCys, pAsp, and pGlu, it is catastrophic.
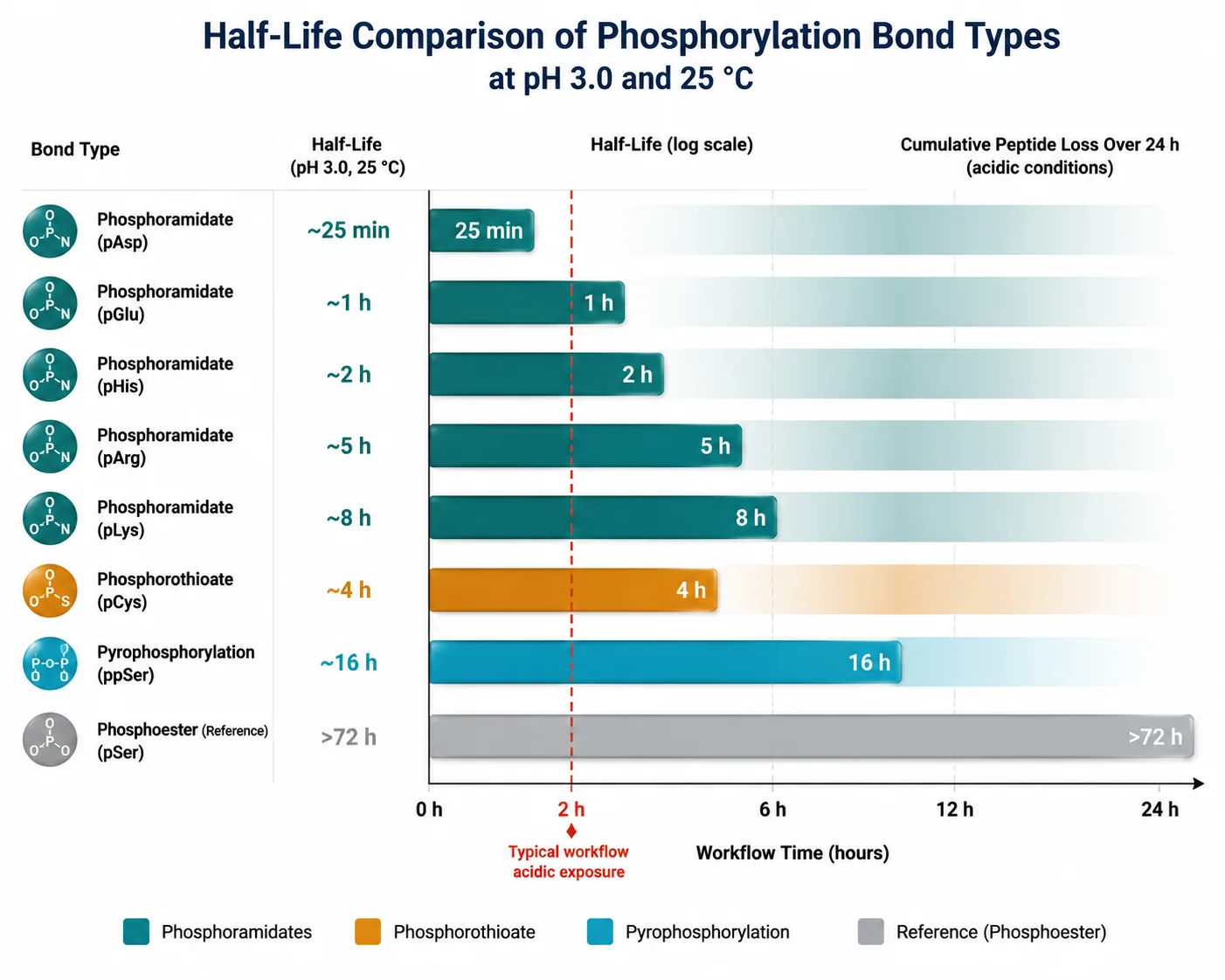
A peptide carrying pHis loaded onto a TiO₂ column at pH 2.7 in 0.1% TFA loses a significant fraction of its phosphate modification before the LC gradient starts. What reaches the mass spectrometer is a mixture of the phosphorylated and dephosphorylated forms, diluting the signal and complicating quantification. For pAsp and pGlu, the loss approaches completeness under standard conditions, explaining their near-total absence from conventional phosphoproteomic datasets.
Biological Importance
Bacterial Two-Component Systems
The biological significance of non-canonical phosphorylation is most firmly established in prokaryotes, where the two-component system (TCS) — a sensor histidine kinase paired with a response regulator — constitutes the dominant signal transduction architecture. The sensor kinase autophosphorylates on a conserved histidine residue, then transfers the phosphoryl group to an aspartate residue on the cognate response regulator. This single pair of non-canonical modifications — pHis and pAsp — controls chemotaxis, osmoregulation, nutrient sensing, antibiotic resistance, and virulence factor expression across the bacterial kingdom. A typical bacterial genome encodes 30–70 TCS pairs, meaning pHis and pAsp are not rare modifications in bacteria; they are the core biochemical language of environmental sensing.
Mammalian Histidine Phosphorylation
In mammals, the NME (non-metastatic) family — NME1 (NM23-H1) and NME2 (NM23-H2) — are the best-characterized histidine kinases. NME1 phosphorylates histidine on the KCa3.1 potassium channel (pHis358), regulating channel activity; on the C-terminal tail of Dynamin-1, influencing endocytosis; and on histone H4 (pHis18 and pHis75), potentially linking histidine phosphorylation to chromatin regulation. The phosphatases PHPT1 (phosphohistidine phosphatase 1) and LHPP (phospholysine phosphohistidine inorganic pyrophosphate phosphatase) remove phosphate from pHis, and their dysregulation has been associated with tumor progression — LHPP is downregulated in hepatocellular carcinoma, and its loss increases total cellular pHis levels and promotes proliferation.
Beyond NME1/2, histidine phosphorylation sites in the tens of thousands have recently been catalogued in human proteomes using UPAX-based enrichment, spanning diverse functional categories from RNA binding and splicing to cytoskeletal regulation. The sheer breadth of the emerging pHis proteome suggests that histidine phosphorylation is not a niche signaling curiosity in mammals but a regulatory layer of comparable breadth to tyrosine phosphorylation — and one that has been essentially invisible to standard methods until very recently.
Pyrophosphorylation and Cellular Stress
The over 700 ppSer sites identified in human cells are disproportionately found on proteins involved in ribosome biogenesis, RNA processing, and transcription. The non-enzymatic installation mechanism — driven by the high-energy diphosphate group — suggests ppSer may serve as a sensor of cellular energy status, marking proteins in response to metabolic flux. The functional consequences of ppSer remain incompletely understood, but the technology to detect and quantify ppSer site-specifically has only existed for a few years.
Among the proteins carrying ppSer, nucleolin — a multifunctional nucleolar phosphoprotein involved in rRNA transcription and ribosome assembly — harbors multiple ppSer sites within its acidic N-terminal domain, a region rich in runs of consecutive serines and glutamates that form an extended negatively charged surface. SR-family splicing factors, including SRSF1 and SRSF2, also carry ppSer modifications in their arginine-serine-rich (RS) domains, raising the possibility that pyrophosphorylation modulates spliceosome assembly by altering the charge density that drives protein-RNA interactions. The non-enzymatic installation mechanism — dependent on local diphosphate concentration and facilitated by the acidic amino acid context that positions a nucleophilic serine hydroxyl adjacent to a high-energy pyrophosphate donor — suggests ppSer levels may fluctuate with cellular ATP hydrolysis rates, positioning ppSer as a potential direct sensor of energy status rather than a kinase-driven signaling event. This hypothesis, if validated, would place ppSer in a category distinct from all other phosphorylation: a metabolically-installed mark that bypasses the kinase-phosphatase paradigm entirely.
The field is still in a discovery phase, and Creative Proteomics offers proteomic PTM characterization for researchers investigating non-canonical modifications at scale.
Why Standard Phosphoproteomics Fails
The conventional phosphoproteomics enrichment workflow — immobilized metal affinity chromatography (IMAC) with Fe³⁺ or Ga³⁺, or titanium dioxide (TiO₂) — operates at pH 2.5–3.0 in the loading and wash buffers. At this pH, phosphate groups on pSer, pThr, and pTyr are partially protonated, enabling specific binding to the metal or metal oxide surface. But the same pH regime that creates binding specificity for the phosphate moiety simultaneously hydrolyzes the P–N, P–S, and acyl-phosphate bonds of non-canonical modifications.
This is not a fixable problem within the metal-affinity paradigm. The binding chemistry and the hydrolysis chemistry are coupled — you cannot operate at a pH that spares pHis and pAsp while maintaining the phosphate-specific binding that makes IMAC and TiO₂ work. Raising the enrichment pH to 6.0–7.0, where non-canonical phosphates are more stable, collapses selectivity: carboxylic acid side chains (Asp, Glu) and the peptide C-terminus become deprotonated and compete for metal binding, and the phosphate group on pSer/pThr becomes doubly deprotonated and binds with lower specificity. The result is a contaminated enrichment with poor phosphopeptide recovery.
This inherent chemical conflict means that accessing non-canonical phosphorylation requires abandoning the metal-affinity principle entirely and adopting chemically orthogonal enrichment strategies. For researchers evaluating where to invest method development effort, Creative Proteomics offers PTM analysis method development spanning both canonical and non-canonical phosphorylation workflows.
Enrichment Strategies
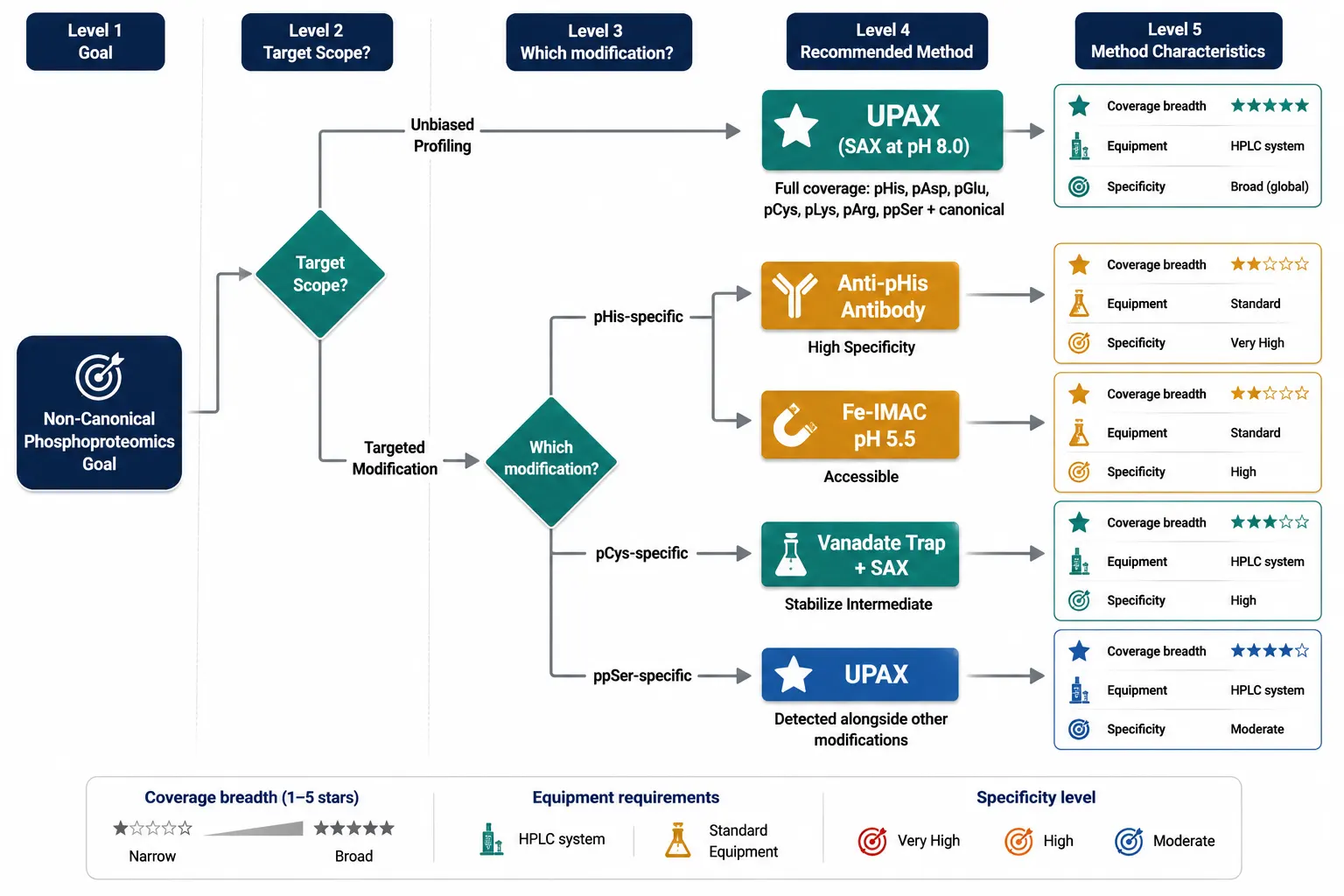
The past five years have produced a set of enrichment methods that operate at neutral-to-basic pH, circumventing the acid lability problem. No single method captures all non-canonical modifications with equal efficiency; the choice depends on which modification class is the primary target.
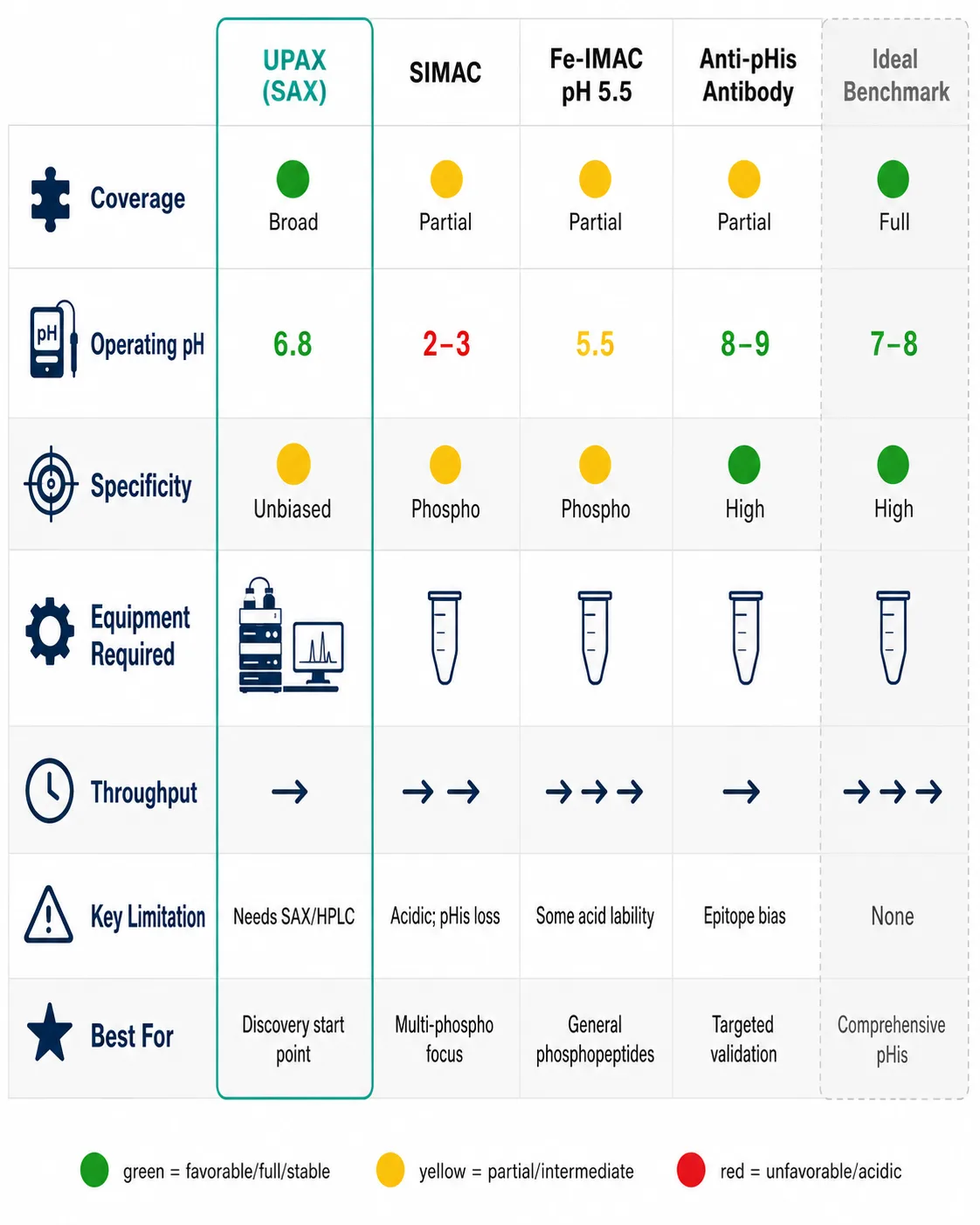
UPAX: Strong Anion Exchange at Neutral pH
The 2024 UPAX (universal phosphoproteomics) method represents the most significant advance in non-canonical phosphopeptide enrichment. The principle is deceptively simple: replace metal-affinity binding at acidic pH with strong anion exchange chromatography at pH 8.0. At this pH, phosphate groups — regardless of the amino acid they are attached to — carry a −2 charge and bind strongly to quaternary ammonium-functionalized SAX resin through electrostatic interaction. Phosphopeptides are eluted with a high-salt step gradient, and the eluate is desalted and analyzed by LC-MS/MS.
The method captures pSer, pThr, pTyr, pHis, pAsp, pGlu, pLys, pArg, and pCys simultaneously — the full spectrum — because the binding mechanism recognizes the phosphate group's charge state, not its chemical context. The pH 8.0 operating condition preserves all classes of non-canonical phosphorylation throughout the enrichment. The 2024 study applying UPAX to human cell lysates identified approximately 7,000 unique non-canonical phosphosites, a number that dwarfs all prior non-canonical phosphoproteomic datasets combined.
UPAX has limitations. The SAX enrichment is less specific than IMAC or TiO₂ — acidic non-phosphorylated peptides co-enrich at higher rates, requiring either fractionation or stringent data filtering. The method requires careful pH control; the mobile phases must be freshly prepared, and the column must be equilibrated thoroughly. The salt elution introduces non-volatile salts (NaCl or KCl) that must be removed by desalting before LC-MS, adding a step where acidic conditions could theoretically be reintroduced. Despite these caveats, UPAX is currently the method of choice for unbiased non-canonical phosphoproteomics, and its adoption is accelerating rapidly.
SIMAC: Sequential Elution IMAC
SIMAC (sequential elution from IMAC) partially addresses non-canonical phosphorylation by splitting the IMAC elution into two fractions: mono-phosphorylated peptides are eluted first at acidic pH, and multi-phosphorylated peptides are eluted at basic pH (pH 11.5). The basic elution step preserves some pHis-containing peptides that would be lost in a standard IMAC workflow, though the acidic loading step still results in partial hydrolysis. SIMAC is better than standard IMAC for pHis but does not match UPAX for coverage breadth.
Fe-IMAC at Near-Neutral pH
A modified Fe³⁺-IMAC protocol operates at pH 5.0–5.5 rather than the standard 2.5–3.0. At this intermediate pH, the binding specificity for phosphate is partially retained while acid-catalyzed hydrolysis of P–N bonds is slowed. The trade-off is reduced enrichment specificity — more non-phosphorylated acidic peptides are co-enriched — and the method is effective for pHis but less so for the more acid-labile pAsp and pGlu. Fe-IMAC at pH 5.5 can be useful when SAX-capable HPLC equipment is not available, and it can be implemented on standard IMAC hardware with buffer adjustments.
Anti-pHis Antibody Enrichment
Monoclonal antibodies recognizing 1-phosphohistidine and 3-phosphohistidine are commercially available and have been used for immunoaffinity enrichment of pHis peptides from complex digests. The antibody approach offers high specificity — the false discovery rate from immunoaffinity enrichment is inherently lower than from chemical enrichment because the antibody discriminates pHis from other phosphate modifications. The critical limitation is narrow scope: anti-pHis antibodies do not recognize pLys, pArg, pAsp, pGlu, pCys, or ppSer. An anti-pHis enrichment experiment answers the question "what histidine phosphorylation sites are present?" — but it cannot answer "what non-canonical phosphorylation sites are present?" For a comprehensive view, UPAX is the starting point; antibody-based methods are best deployed for targeted follow-up or validation.
For researchers who have identified candidate non-canonical phosphosites and need sequence-level confirmation, Creative Proteomics provides de novo peptide sequencing and top-down PTM characterization for modification site mapping at amino acid resolution.
MS Detection
Enrichment gets non-canonical phosphopeptides to the mass spectrometer. Fragmentation determines whether you can identify them.
The standard collision-induced dissociation (CID) and higher-energy collisional dissociation (HCD) used in most phosphoproteomics experiments preferentially cleave the phosphate group from the peptide backbone before peptide bonds fragment. For pSer and pThr, this neutral loss of H₃PO₄ (−98 Da) is well-characterized and can be exploited for targeted detection. For non-canonical phosphorylation, the fragmentation behavior is different and, in some cases, diagnostically useful.
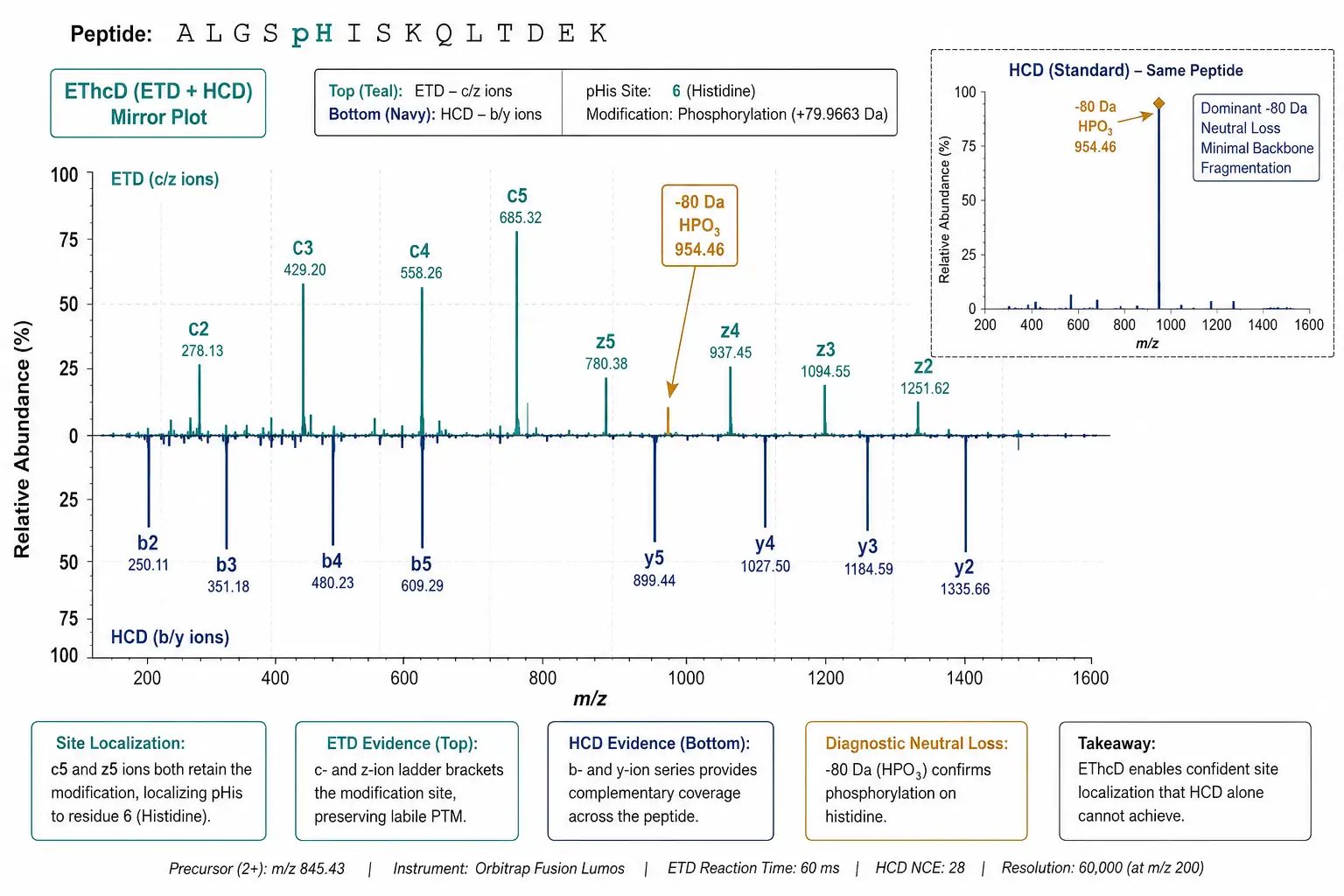
EThcD: The Preferred Fragmentation Mode
Electron transfer with supplemental higher-energy collisional dissociation (EThcD) combines electron transfer dissociation (ETD), which preserves labile modifications during fragmentation, with HCD, which generates peptide backbone fragments. For pHis-containing peptides, EThcD produces sequence-informative c- and z-ions from ETD alongside b- and y-ions from HCD, maximizing sequence coverage while retaining the phosphate on the histidine residue. This is critical because a CID or HCD spectrum showing neutral loss of phosphate without sequence ions at the modified residue cannot localize the modification site. EThcD has become the standard fragmentation mode for non-canonical phosphoproteomics on Orbitrap instruments equipped with ETD capability (Fusion Lumos, Eclipse, and Tribrid series).
Neutral Loss Signatures
Each non-canonical modification class exhibits characteristic neutral loss behavior that can aid identification:
- pHis: neutral loss of HPO₃ (−80 Da) is the dominant pathway under CID/HCD, distinct from the −98 Da loss of H₃PO₄ from pSer/pThr. This −80 Da loss is diagnostic for N-phosphorylation.
- pCys: loss of H₃PO₄ (−98 Da), similar to pSer/pThr, making pCys difficult to distinguish from canonical phosphorylation by fragmentation alone. Sequence context (presence of Cys at the site) is essential.
- pAsp/pGlu: the acyl-phosphate bond fragments readily, producing dominant neutral loss of HPO₃ (−80 Da) or H₃PO₄ (−98 Da) depending on sequence context, often with limited backbone fragmentation. EThcD is essential for confident site assignment.
- ppSer: loss of H₄P₂O₇ (−178 Da) and H₃PO₄ (−98 Da) produce a dual neutral-loss pattern that is characteristic of pyrophosphorylation.
Database Search Configuration
Standard phosphoproteomics database searches configured for S/T/Y phosphorylation will not identify non-canonical modifications. The search must be configured with variable modifications on the target residues: phosphorylation on H, C, D, E, K, and R, in addition to the canonical S/T/Y. This increases the search space considerably — adding six residue types with a variable modification multiplies the number of theoretical candidates — and requires careful false discovery rate (FDR) control. A common strategy is to run an initial broad search with all modifications enabled at 1% FDR at the PSM level, then apply a site localization probability filter (≥0.75 using a tool such as PhosphoRS or Ascore) to remove ambiguous assignments. Given the higher false localization rate for non-canonical modifications, sites passing only the FDR filter without the localization filter should be treated as tentative until validated.
For quantitative non-canonical phosphoproteomics, Creative Proteomics provides protein identification services with high-resolution MS/MS and custom modification search configuration.
Tour by Residue
Phosphohistidine (pHis)
The most studied non-canonical modification. Two isoforms (1-pHis and 3-pHis) with distinct thermodynamic stabilities and antibody recognition profiles. Mammalian writers: NME1, NME2. Mammalian erasers: PHPT1, LHPP, PGAM5. Best detected by UPAX enrichment with EThcD fragmentation. Anti-pHis antibodies available for immunoaffinity enrichment and western blot validation. Key biological contexts: tumor suppression (LHPP loss), ion channel regulation (KCa3.1), chromatin (histone H4), endocytosis (Dynamin-1).
Phosphoaspartate (pAsp)
Half-life at pH 3.0 frequently under 30 minutes, making it the most challenging non-canonical modification to detect. In the bacterial chemotaxis system, the CheY response regulator receives a phosphoryl group from the sensor kinase CheA onto Asp57; phosphorylation induces a conformational shift in the β4-α4 loop that reorients the CheY FliM-binding surface, switching flagellar rotation from counterclockwise (smooth swimming) to clockwise (tumbling). In P-type ATPases, phosphoaspartate is not a regulatory modification but a catalytic obligate: the Ca²⁺-ATPase (SERCA) autophosphorylates on Asp351 during the E1→E1P transition, using the energy of the acyl-phosphate bond to drive the E1P→E2P conformational change that translocates two calcium ions across the sarcoplasmic reticulum membrane. Detection requires SAX at neutral pH with minimal exposure to acidic conditions at any workflow step. The limited mammalian pAsp literature outside of P-type ATPases partly reflects detection difficulty rather than biological absence.
Phosphoglutamate (pGlu)
Similar chemistry to pAsp with slightly longer acid half-lives owing to the additional methylene group in the glutamate side chain, which reduces the electrophilicity of the carbonyl carbon slightly relative to aspartate. In the bacterial phosphoenolpyruvate:sugar phosphotransferase system (PTS), the glucose-specific Enzyme II permease undergoes phosphorylation on a conserved glutamate residue as part of the phosphoryl relay that drives sugar import. Beyond the PTS, pGlu has been detected in mammalian cell extracts by UPAX-based enrichment, though the site counts are substantially lower than for pHis, and no dedicated mammalian pGlu kinase or phosphatase has been confirmed. Research into mammalian pGlu biology remains nascent, likely in part due to the extreme acid lability of the acyl-phosphate bond.
Phospholysine (pLys) and Phosphoarginine (pArg)
Both phosphoramidates with stability profiles similar to pHis. pLys detected on histones; pArg less well characterized in mammalian systems. Neither has commercial antibodies available, making SAX-based methods the primary detection route. The bacterial phosphotransferase system (PTS) uses a phosphohistidine-to-phosphocysteine relay in the glucose-specific permease, showcasing the biological coupling between different non-canonical phosphorylation chemistries.
Phosphocysteine (pCys)
Distinguished by its P–S bond chemistry and occurrence in PTP catalytic intermediates. Detection requires trapping with vanadate or tungstate to stabilize the normally transient modification. The presence of pCys in non-catalytic regulatory sites remains an open question — one that improved detection methods may soon answer.
Pyrophosphoserine (ppSer)
Over 700 sites catalogued. Enriched on acidic serine-rich motifs. Non-enzymatic installation linked to metabolic state. Functional consequences under investigation. First detected by fortuitous observation of the −178 Da neutral loss signature. ppSer sites can be detected by UPAX alongside other non-canonical modifications.
Kinases and Phosphatases
The kinase and phosphatase landscape for non-canonical phosphorylation illustrates how much of this signaling layer remains unmapped. For canonical phosphorylation, approximately 500 human protein kinases and approximately 200 human protein phosphatases have been catalogued. For non-canonical phosphorylation, the confirmed writer and eraser enzymes can be counted on two hands.
The mammalian histidine kinase family currently includes NME1, NME2, and potentially NME3 and NME4, though the latter remain less well characterized. The histidine phosphatases PHPT1, LHPP, and PGAM5 have confirmed pHis substrates, and LHPP's tumor-suppressive role in hepatocellular carcinoma makes it the most clinically relevant non-canonical phosphatase identified to date.
For pAsp and pGlu, no dedicated mammalian kinases or phosphatases are currently confirmed, though the Ca²⁺-ATPase undergoes autophosphorylation on Asp351 during its catalytic cycle. For pCys, the modification is installed by the PTPs themselves as a catalytic intermediate rather than by a dedicated kinase. For ppSer, the non-enzymatic installation mechanism raises the question of whether dedicated ppSer kinases exist at all.
The concept of "kinase targeting" — identifying which kinase phosphorylates a given site — extends to non-canonical phosphorylation but with far fewer tools. Chemical biology approaches, including ATP analog-sensitive kinase engineering and photocrosslinking probes, are being adapted for non-canonical kinases. For a broader discussion of chemoproteomic approaches to kinase characterization, see our guide on chemoproteomics for covalent inhibitor development and cysteine reactivity profiling. For interaction mapping of non-canonical kinase-substrate pairs, Creative Proteomics offers protein-protein interaction analysis with quantitative MS readout — see also our guide on AP-MS for protein interaction mapping from pull-down to identification.
Data Analysis Challenges
Non-canonical phosphoproteomic data presents analytical challenges beyond those of canonical phosphoproteomics, and canonical phosphoproteomics is already among the most computationally demanding proteomic workflows.
The expanded search space created by adding six variable modification types (H, C, D, E, K, R) to the canonical three (S, T, Y) increases the number of candidate PSMs exponentially. At a fixed 1% FDR threshold, a larger search space means more false positives survive filtering. A two-tier FDR strategy — 1% at the PSM level followed by a stricter 1% at the site level — is recommended, alongside the site localization probability filter (≥0.75) mentioned earlier.
Site localization is inherently harder for non-canonical modifications than for canonical ones. The diagnostic fragment ions used by PhosphoRS and Ascore to distinguish between adjacent candidate sites were developed and validated on pSer/pThr/pTyr data. Their performance on pHis, pAsp, and pCys — where fragmentation patterns differ and fewer benchmark datasets exist — has not been systematically validated. Until such validation is published, non-canonical phosphosite assignments should be treated as provisional unless supported by orthogonal evidence: synthetic phosphopeptide co-elution, site-directed mutagenesis, or independent enrichment with a different method.
Motif analysis for kinase prediction — a standard downstream step in canonical phosphoproteomics — faces the problem that non-canonical kinase consensus motifs are almost entirely unknown. The motif-x algorithm can extract enriched sequence motifs from a list of non-canonical phosphosites, and the resulting motifs may reveal novel consensus sequences for uncharacterized kinases. However, the sample sizes for individual non-canonical modification types in current datasets rarely reach the hundreds of sites needed for robust motif extraction. For researchers managing large non-canonical phosphoproteomic datasets, Creative Proteomics provides proteomics bioinformatics support including custom database search configuration, site localization scoring, and motif enrichment analysis.
FAQ
What is non-canonical protein phosphorylation?
Non-canonical phosphorylation refers to the attachment of phosphate groups to amino acid residues other than serine, threonine, and tyrosine — primarily histidine (pHis), aspartate (pAsp), glutamate (pGlu), cysteine (pCys), lysine (pLys), and arginine (pArg), plus pyrophosphorylation of serine (ppSer). These modifications use different chemical bonds (P–N, P–S, acyl-phosphate) than the P–O ester bonds of canonical phosphorylation.
Why are non-canonical phosphorylation sites missed in standard phosphoproteomics?
The P–N, P–S, and acyl-phosphate bonds in non-canonical modifications are acid-labile and hydrolyze at the low pH (2.5–3.0) used in standard TiO₂/IMAC enrichment workflows. The modifications are destroyed before they reach the mass spectrometer.
What is the best enrichment method for non-canonical phosphoproteomics?
UPAX (strong anion exchange at pH 8.0), published in 2024, is the current method of choice. It captures the full spectrum of canonical and non-canonical phosphopeptides simultaneously because the binding mechanism depends on phosphate charge state, not chemical context. Anti-pHis antibodies offer high specificity for targeted pHis analysis, and Fe-IMAC at pH 5.5 is an accessible alternative when SAX equipment is not available.
How many non-canonical phosphorylation sites exist in human cells?
The 2024 UPAX study identified approximately 7,000 unique non-canonical phosphosites in human cells under basal conditions — approximately one-third the number of canonical phosphosites. This number is expected to grow as enrichment methods improve.
Which enzymes write and erase non-canonical phosphorylation?
Confirmed mammalian histidine kinases: NME1, NME2. Histidine phosphatases: PHPT1, LHPP, PGAM5. For pAsp, pGlu, and pCys, dedicated mammalian kinases and phosphatases remain largely uncharacterized. ppSer appears to be installed non-enzymatically.
Can I use standard phosphoproteomics software for non-canonical data?
Yes, but the search must be configured with variable modifications on H, C, D, E, K, and R in addition to S/T/Y. Site localization tools (PhosphoRS, Ascore) were validated on canonical data and should be used with caution for non-canonical site assignment. A localization probability threshold of ≥0.75 is recommended.
What fragmentation mode is best for non-canonical phosphopeptides?
EThcD on Orbitrap Tribrid instruments is the preferred mode. It preserves labile modifications during ETD while providing backbone fragments from HCD, maximizing sequence coverage and site localization confidence. The −80 Da neutral loss of HPO₃ under CID/HCD is diagnostic for N-phosphorylation (pHis, pLys, pArg).
Is non-canonical phosphorylation biologically important in mammals?
Yes. Histidine phosphorylation regulates ion channels (KCa3.1), endocytosis (Dynamin-1), and chromatin (histone H4). LHPP, a histidine phosphatase, is a tumor suppressor in hepatocellular carcinoma. The full regulatory significance is still being uncovered — largely because the tools to detect these modifications only recently became available.
References:
- Houles T, Pansieri C, Roux PP. The expanding landscape of canonical and non-canonical protein phosphorylation. Trends in Biochemical Sciences, 2024, 49(11):986-999. doi:10.1016/j.tibs.2024.08.003.
- Jiang Y, Li Y, Zhang Y, et al. Progress in enrichment methods for protein N-phosphorylation. Chinese Journal of Chromatography, 2024, 42(7):623-631. doi:10.3724/SP.J.1123.2024.03004.
- Hardman G, Perkins S, Brownridge PJ, et al. Strong anion exchange-mediated phosphoproteomics reveals extensive human non-canonical phosphorylation. EMBO Journal. 2019;38(21):e100847. doi:10.15252/embj.2018100847.
- Zhang Y, Li J, Wang H, et al. Advances in protein phosphorylation analysis: from canonical to non-canonical targets. International Journal of Molecular Sciences. 2024;25(17):9365. doi:10.3390/ijms25179365.
- Marmelstein AM, Moreno J, Fiedler D. Chemical approaches to studying labile amino acid phosphorylation. Topics in Current Chemistry. 2017;375:22. doi:10.1038/s41589-023-01465-5.
- Marmelstein AM, Moreno J, Fiedler D. Chemical Approaches to Studying Labile Amino Acid Phosphorylation. Topics in Current Chemistry, 2017, 375:22. doi:10.1007/s41061-017-0111-1.













