Think of a protein as a lock, and a drug as a key. Most drugs work by temporarily jamming themselves into the lock — they bind, they unbind, and the protein goes back to doing whatever it was doing. Covalent inhibitors are different. They don't just sit in the lock; they weld themselves to it. Once attached, they stay attached — permanently. This permanent bonding is what makes covalent drugs so powerful: they can shut down a disease-driving protein completely, and the effect lasts until the cell makes new copies of that protein.
But how do you find the right cysteine — the right spot on the right protein — to weld your drug to? And once you've found it, how do you prove your drug is hitting that spot and not welding itself to hundreds of other proteins by accident? This is the problem that chemoproteomics, and specifically activity-based protein profiling (ABPP), was invented to solve.
This article explains how ABPP works, how it drives covalent inhibitor discovery from fragment screening to clinical candidate, and why the technology is now expanding beyond its traditional focus on cysteine to embrace lysine, tyrosine, and other amino acids as covalent targets.
What Is Chemoproteomics?
Chemoproteomics — sometimes called chemical proteomics — is the use of small-molecule probes to map protein function, interactions, and drug binding across the entire proteome in a single experiment. Unlike traditional biochemistry, which studies one protein at a time in a test tube, chemoproteomics asks a much bigger question: "When I add this drug-like molecule to a living system, which proteins does it touch?" Creative Proteomics' Proteomics Service provides the analytical infrastructure — from sample preparation through quantitative LC-MS/MS — that makes chemoproteomics experiments possible at scale.
The core tool of chemoproteomics is the activity-based probe. An activity-based probe has three parts, which you can visualize like a three-component delivery system. The warhead is the business end — a reactive chemical group that forms a covalent bond with the target amino acid on a protein. The linker is a spacer that connects the warhead to the tag. And the tag is what lets you fish out the labeled proteins later — traditionally a biotin molecule that acts like a molecular hook, or more commonly today, an alkyne group that serves as a chemical handle for attaching a reporter after the labeling step.
 Figure 1: ABPP Probe Architecture — The Three-Component Design
Figure 1: ABPP Probe Architecture — The Three-Component Design
A modern isometric 3D scientific illustration showing the three-component architecture of an activity-based protein profiling (ABPP) probe: a cysteine-reactive warhead covalently attached to a protein surface, a flexible polyethylene glycol linker chain, and an alkyne handle for copper-catalyzed azide-alkyne cycloaddition (CuAAC) attaching a biotin tag.
Activity-Based Protein Profiling: The Complete Workflow
An ABPP experiment follows four logical stages: label, enrich, digest, and identify. Understanding each stage is essential because the choices made at each one determine what kinds of targets you find and how confident you can be in the results.
Stage 1: Probe Labeling
You expose your biological sample — typically a cell lysate, or in more advanced setups, intact living cells — to an activity-based probe carrying a cysteine-reactive warhead. The most widely used warhead is iodoacetamide-alkyne (IA-alkyne). The iodoacetamide portion reacts selectively with the thiol (-SH) group of cysteine residues to form a stable thioether bond, while the alkyne portion waits silently for the next step.
The labeling conditions matter enormously. Probe concentration, incubation time, pH, and whether you work in lysate or live cells all influence which cysteines get labeled and to what extent. In lysate-based experiments, you get broad coverage because the probe has unrestricted access to proteins. But you also lose the native cellular environment — protein complexes, membranes, and endogenous metabolites that may shield certain cysteines in the living cell. Live-cell labeling with cell-permeable probes preserves this native context but introduces its own challenges: the probe must cross the plasma membrane, distribute evenly through cellular compartments, and avoid triggering toxicity responses before labeling is complete.
Stage 2: Enrichment
After labeling, you break open the cells (if you haven't already), denature the proteins, and use click chemistry to attach a biotin tag to the alkyne handle on your probe. Click chemistry — specifically the copper-catalyzed azide-alkyne cycloaddition (CuAAC) — is the enabling reaction that makes ABPP practical. It is fast, selective, and works in complex biological mixtures. Once biotin is attached, you capture the probe-labeled proteins on streptavidin beads — streptavidin binds biotin with one of the strongest non-covalent interactions in biology.
Stage 3: On-Bead Digestion
The captured proteins are washed extensively to remove non-specifically bound material, then digested into peptides using trypsin. This step converts full-length proteins into a mixture of short peptides suitable for mass spectrometry analysis.
Stage 4: LC-MS/MS Identification and Quantification
The peptide mixture is separated by liquid chromatography and analyzed by tandem mass spectrometry (MS/MS). Each peptide is fragmented in the mass spectrometer, and the resulting fragment ion spectrum is matched against a protein sequence database to identify which proteins were present — and, critically, which specific cysteine residues carried the probe modification. Creative Proteomics offers Protein Identification Services that provide the site-level resolution essential for ABPP workflows, distinguishing covalent probe-labeled peptides from background. This site-level resolution is what distinguishes ABPP from simpler pull-down approaches.
The Warhead Toolbox: Cysteine and Beyond
Cysteine has dominated covalent drug discovery for a simple chemical reason: its thiol side chain is the most intrinsically nucleophilic group found in proteins. At physiological pH, the thiolate anion (-S-) can attack electron-deficient carbons in acrylamides, chloroacetamides, and other electrophilic warheads with remarkable efficiency. But not all cysteines are created equal. The reactivity of a given cysteine depends on its local protein environment — nearby positive charges that stabilize the thiolate, solvent accessibility, and the pKa of the thiol group, which can vary by several pH units depending on the surrounding residues.
The major warhead classes, ranked by their prevalence in covalent drug discovery, include:
Acrylamides are the most successful warhead class, used in the approved drugs ibrutinib (targeting BTK), osimertinib (targeting EGFR T790M), and sotorasib (targeting KRAS G12C). They react via a Michael addition mechanism: the cysteine thiolate attacks the terminal carbon of the alpha,beta-unsaturated carbonyl, forming a covalent adduct. Acrylamides are relatively inert toward free glutathione, which means they have low off-target reactivity — a key advantage for drug development.
Chloroacetamides, including the IA-alkyne probe used in most ABPP experiments, are more reactive than acrylamides and label a broader swath of the cysteinome. This higher reactivity makes them excellent for discovery — you want to see as many ligandable cysteines as possible — but less ideal for therapeutic molecules, where selectivity is paramount.
Sulfonyl fluorides and related sulfur(VI) fluoride exchange (SuFEx) warheads react with cysteine but also with lysine, tyrosine, and histidine, making them versatile but less selective. Vinyl sulfones and electron-deficient alkynes round out the commonly used cysteine-directed warheads.
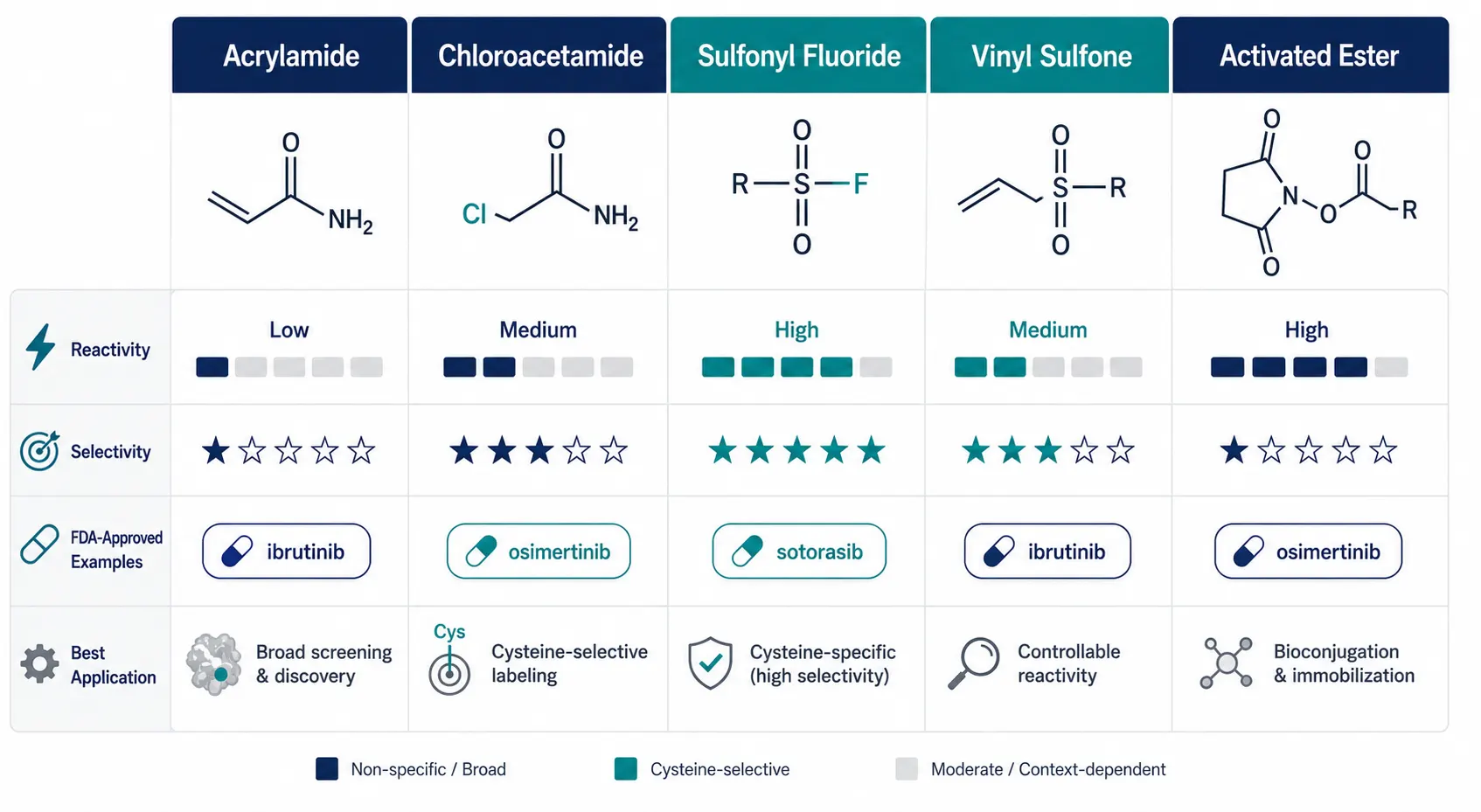 Figure 2: Covalent Warhead Class Comparison Matrix
Figure 2: Covalent Warhead Class Comparison Matrix
A modern flat design comparison matrix presenting five major covalent warhead classes — acrylamide, chloroacetamide, sulfonyl fluoride, vinyl sulfone, and activated ester — with comparison data on reactivity, selectivity, FDA-approved examples, and best application for each class.
Beyond Cysteine: The Next Frontier
The past three years have seen an explosion of interest in targeting amino acids other than cysteine. Lysine, with its nucleophilic epsilon-amino group, can be targeted with activated esters (N-hydroxysuccinimidyl esters, or STP esters) and dichlorotriazines — Creative Proteomics provides Proteomic Analysis of Post-translational Modifications Service for comprehensive PTM characterization relevant to covalent ligand development. Tyrosine can be engaged through sulfur-triazole exchange (SuTEx) chemistry and aryl fluorosulfates. Histidine, methionine, aspartate, glutamate, and even arginine have all yielded to carefully designed electrophilic warheads, dramatically expanding the fraction of the proteome considered "covalently ligandable." A systematic 2025 study (Zhou et al., Biomolecules 2025) profiled diverse electrophilic probes against the entire proteome and identified high-selectivity probes for nine different amino acids plus the protein N-terminus.
Quantitative Platforms: isoTOP-ABPP, TMT-ABPP, and Beyond
Identifying which proteins carry a probe-labeled cysteine tells you which cysteines are reactive. But in drug discovery, the more important question is: when I add my drug candidate, which cysteines are blocked from reacting with the probe? This competition experiment is the heart of covalent inhibitor target identification.
isoTOP-ABPP (isotopic Tandem Orthogonal Proteolysis-ABPP) is the classic platform for this competitive profiling. In a typical isoTOP-ABPP experiment, you split a proteome sample into two aliquots. One is treated with your covalent inhibitor of interest; the other receives a vehicle (DMSO) control. Both are then labeled with an IA-alkyne probe. The probe will label all accessible reactive cysteines, but in the inhibitor-treated sample, the cysteines already occupied by your inhibitor are protected — they can't react with the probe. After click chemistry with isotopically differentiated (light vs. heavy) cleavable biotin tags, enrichment, and MS analysis, you compare the light/heavy ratio for each cysteine. A high ratio means the cysteine was strongly competed by your inhibitor, identifying it as a direct target.
The key strength of isoTOP-ABPP is its quantitative precision — the light/heavy ratio provides a direct measurement of target engagement. For competitive ABPP experiments that do not require isotopic labeling, Creative Proteomics' Label-free Quantification service offers an accessible alternative for initial target engagement screening. Its limitation is throughput: traditional isoTOP-ABPP compares only two conditions per experiment.
TMT-ABPP (Tandem Mass Tag-ABPP) addresses the throughput limitation by using isobaric mass tags that allow multiplexing of up to 18 samples in a single MS run — Creative Proteomics offers TMT based proteomics service with optimized reporter ion acquisition parameters designed to mitigate ratio compression artifacts. Instead of comparing just inhibitor vs. vehicle, you can simultaneously profile dose-response curves (multiple inhibitor concentrations), time courses, or panels of structurally related compounds. The TMT reagents carry different isotopic distributions that produce distinct reporter ions upon MS/MS fragmentation, enabling relative quantification across all channels simultaneously. The trade-off is that TMT-based quantification can suffer from ratio compression due to co-isolated interfering peptides, requiring careful experimental design and MS acquisition parameters.
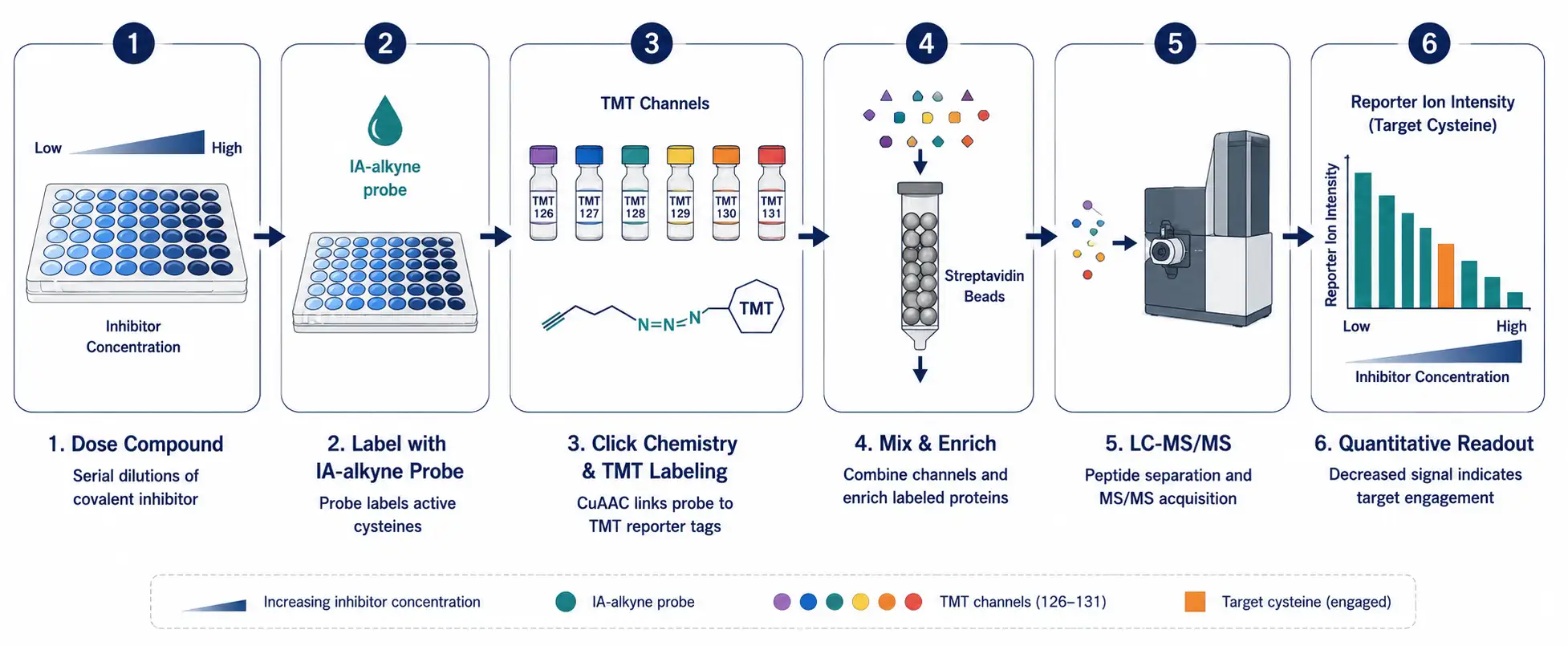 Figure 3: TMT-ABPP Competitive Profiling Workflow
Figure 3: TMT-ABPP Competitive Profiling Workflow
A horizontal scientific workflow diagram showing the TMT-ABPP competitive profiling pipeline in six stages: dose compound, label with IA-alkyne probe, click chemistry and TMT labeling, mix and enrich on streptavidin beads, LC-MS/MS analysis, and quantitative readout as a bar chart of decreasing reporter ion intensity.
A more recent innovation is isoDTB-ABPP, which replaces the cleavable biotin tag with a desthiobiotin group that can be eluted with organic solvent rather than proteolytic cleavage. This streamlines the workflow and reduces sample handling losses. Combined with the FragPipe computational platform for chemoproteomics data analysis — supported by Creative Proteomics' Bioinformatics for Proteomics services for customized chemoproteomics data processing — isoDTB-ABPP has become a high-performance pipeline for both academic and industry labs.
Cysteine Reactivity Profiling: From Fragment Screening to Lead Optimization
Covalent fragment screening differs fundamentally from traditional high-throughput screening. Traditional HTS tests millions of pre-optimized drug-sized molecules (MW ~400-500 Da) at low concentrations. Covalent fragment-based screening tests a few hundred to a few thousand very small electrophilic fragments (MW ~150-250 Da) at higher concentrations, then uses MS-based ABPP to read out which proteins — and which specific cysteines — each fragment engages. Creative Proteomics' Shotgun Protein Identification service enables the proteome-wide detection required for fragment-based cysteine ligandability mapping.
The logic is compelling: fragments are small enough to sample a much larger fraction of the proteome's ligandable space. Each fragment typically engages a handful of cysteine sites rather than a single target. By screening a library of, say, 500 acrylamide fragments against a cancer cell proteome, you might identify 2,000-3,000 ligandable cysteines across 1,500 proteins — a map of the "ligandable cysteinome" that can be mined for years.
Hit triage is the critical step. Not every cysteine that binds a fragment is worth pursuing. A good covalent target site should be: present in a protein relevant to your disease biology, not conserved in anti-targets where inhibition would cause toxicity, and located in a functional region of the protein (active site, allosteric pocket, or protein-protein interaction interface) where covalent modification would alter activity. Creative Proteomics' Protein-Protein Interaction Analysis Service can help determine whether a ligandable cysteine resides at a functionally relevant protein-protein interface.
From fragment hit to lead compound, the medicinal chemistry optimization cycle follows familiar principles — improving potency (measured by the kinetic parameter kinact/KI), selectivity (measured by competitive ABPP against the full proteome), and drug-like properties — but with the added dimension of covalent engagement kinetics. A covalent inhibitor's potency depends not only on how tightly it binds (KI) but on how fast it forms the covalent bond (kinact). This means optimization requires tracking both binding affinity and reaction rate simultaneously.
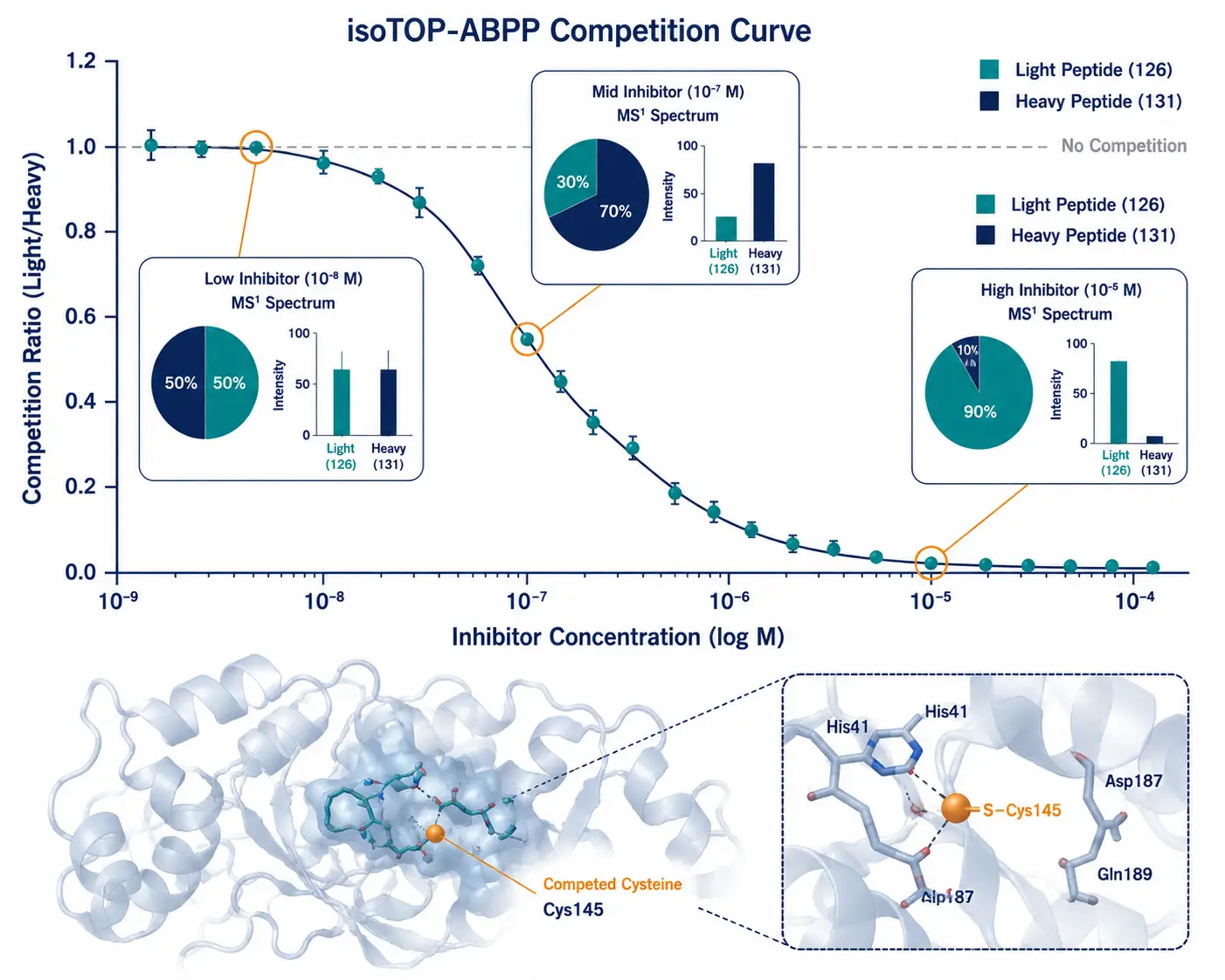 Figure 4: isoTOP-ABPP Target Engagement Dose-Response
Figure 4: isoTOP-ABPP Target Engagement Dose-Response
A scientific data visualization combining a dose-response competition curve (competition ratio vs. inhibitor concentration) with annotated protein structure showing the competed cysteine residue highlighted within a binding pocket, and MS1 spectra insets at selected concentrations.
Case Studies: How ABPP Delivers Clinical Candidates
KRAS G12C and Sotorasib
For decades, KRAS was considered undruggable — its surface lacks deep binding pockets, and it binds GTP so tightly (picomolar affinity) that competitive inhibitors seemed hopeless. The breakthrough came from chemoproteomics. Researchers used disulfide-based fragment screens with MS-based ABPP to identify fragments that bound to a previously unappreciated cryptic pocket near the G12C mutant cysteine. This pocket, now called the switch-II pocket, is only accessible in the GDP-bound (inactive) state of KRAS. By targeting the mutant cysteine in this pocket, sotorasib locks KRAS G12C in its inactive conformation, preventing oncogenic signaling. Sotorasib received FDA approval in 2021 for KRAS G12C-mutant non-small cell lung cancer — a direct product of chemoproteomics-guided covalent drug discovery.
BTK and Ibrutinib
Ibrutinib was developed through medicinal chemistry optimization of a covalent acrylamide warhead targeting Cys481 in the ATP-binding pocket of Bruton's tyrosine kinase (BTK). While ibrutinib itself predates modern proteome-wide ABPP, subsequent chemoproteomic studies revealed its full target profile, including off-target engagement of EGFR, ITK, and TEC family kinases. This off-target information guided the development of next-generation BTK inhibitors (acalabrutinib, zanubrutinib) with improved selectivity profiles validated by ABPP.
NR0B1: An Orphan Nuclear Receptor Becomes Druggable
NR0B1 (also called DAX1) is a nuclear receptor with no known enzymatic activity and no solved crystal structure — the kind of target that traditional drug discovery cannot touch. ABPP screening identified a ligandable cysteine (C274) in NR0B1 that, when covalently modified, destabilizes the NR0B1 protein complex. In KEAP1-mutant non-small cell lung cancer, which depends on NR0B1 for survival, targeting C274 with a covalent ligand suppressed tumor growth. This case illustrates ABPP's unique power: it finds drug-binding sites on proteins that have no business being druggable by conventional criteria.
EGFR and Osimertinib
First-generation EGFR inhibitors (gefitinib, erlotinib) are reversible ATP-competitive inhibitors that target activating mutations (L858R, exon 19 deletions). They are not covalent. The T790M gatekeeper mutation confers resistance. Osimertinib, a covalent acrylamide-based inhibitor targeting Cys797, overcomes T790M resistance while sparing wild-type EGFR — a selectivity profile achieved through careful structure-based design validated by ABPP selectivity profiling.
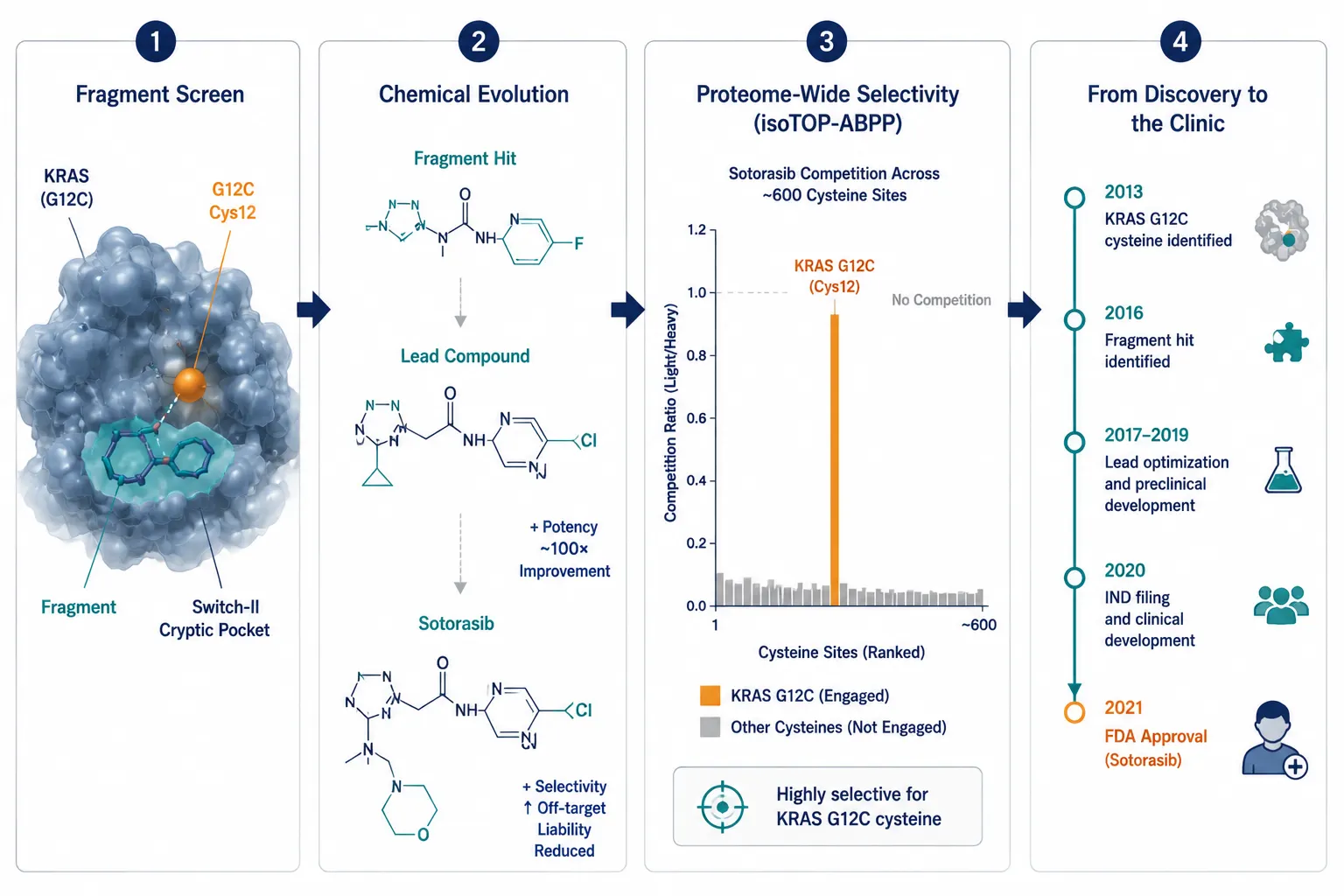 Figure 5: KRAS G12C — From ABPP Fragment Hit to Approved Drug
Figure 5: KRAS G12C — From ABPP Fragment Hit to Approved Drug
A modern case study infographic telling the KRAS G12C drug discovery story in four connected panels: fragment screen identifying cryptic switch-II pocket binder, chemical structure evolution from fragment to sotorasib, isoTOP-ABPP proteome-wide selectivity bar chart, and timeline from cysteine identification to FDA approval.
Electrophilic Fragment Libraries: Design Principles
Building an electrophilic fragment library for covalent screening requires different design principles than building a traditional fragment collection.
First, diversity should span both scaffold diversity and warhead diversity. A typical library includes acrylamides, chloroacetamides, vinyl sulfones, and sulfonyl fluorides attached to structurally diverse scaffolds. Each warhead class has a characteristic reactivity window, so including multiple warheads increases the breadth of the cysteinome you can sample.
Second, fragment reactivity must be calibrated. Fragments that are too reactive will label everything — you get noise, not signal. Fragments that are too inert will label nothing. The ideal fragment has a thiol reactivity (quantified by the glutathione (GSH) half-life assay) in the range of 0.5-50 hours for a 1:1 GSH:fragment ratio at physiological pH. This "Goldilocks zone" ensures sufficient reactivity to engage cysteines while maintaining enough selectivity to produce interpretable ABPP data.
Third, solubility and cell permeability matter early. Many electrophilic fragments are hydrophobic and poorly soluble, limiting their utility in cellular ABPP experiments. Maintaining calculated logP below 3 and including solubilizing groups (tertiary amines, morpholines) in fragment scaffolds improves both solubility and the quality of screening data.
Fourth, libraries should include "clickable" versions of representative fragments. These are fragments equipped with an alkyne handle that allows them to be used directly as ABPP probes for target deconvolution, without needing to synthesize a separate probe molecule.
In Vivo ABPP: Bridging the Gap from Lysate to Animal
A covalent inhibitor that looks exquisitely selective in a cell lysate ABPP experiment can behave very differently in a living animal. Proteins adopt different conformations. Metabolite concentrations (ATP, glutathione, reactive oxygen species) differ dramatically. Drug metabolism can generate reactive metabolites that covalently modify proteins through mechanisms unrelated to the parent compound. These factors mean that in vivo target engagement cannot be reliably predicted from in vitro data alone.
In vivo ABPP attempts to measure target engagement directly in tissues from treated animals. The workflow parallels in vitro ABPP: animals are dosed with the covalent inhibitor, tissues are harvested, homogenized, and labeled ex vivo with an IA-alkyne probe, followed by click chemistry, enrichment, and MS analysis — a workflow supported by Creative Proteomics' Proteomics Service, which provides end-to-end chemoproteomics capabilities from sample preparation to quantitative MS acquisition. The competition readout — which cysteines show reduced probe labeling in drug-treated vs. vehicle-treated animals — identifies in vivo targets.
The challenges are substantial. Drug distribution across tissues is uneven, so target engagement varies by organ. Background labeling is higher in tissue lysates due to abundant endogenous biotinylated proteins and other interfering species. Sample complexity is far greater than in cell culture. And the pharmacokinetic-pharmacodynamic (PK-PD) relationship — how plasma drug concentration relates to the extent and duration of target engagement — must be established to interpret in vivo ABPP data meaningfully.
Despite these challenges, in vivo ABPP has been successfully applied. The KRAS G12C inhibitor ARS-1620 showed time-dependent target engagement in tumor xenografts that correlated with pharmacodynamic biomarkers (p-ERK suppression) and tumor growth inhibition. Similar PK-PD-ABPP correlations have been demonstrated for BTK and EGFR covalent inhibitors. For covalent drug discovery programs, in vivo ABPP target engagement data are increasingly expected by regulators and investors as part of the preclinical package.
The Clinical Covalent Pipeline
The success of covalent inhibitors has transformed the oncology landscape and is now expanding into immunology, neurology, and infectious disease. As of 2025-2026, the FDA has approved over a dozen covalent drugs, with many more in clinical development.
Key clinical-stage programs with strong ABPP foundations include: Vividion Therapeutics' (Bayer) covalent allosteric WRN helicase inhibitor for microsatellite-unstable cancers, discovered through chemoproteomics screening; Jnana Therapeutics' SLC6A19 inhibitor in Phase 3 for phenylketonuria, identified via photoaffinity labeling chemoproteomics; Frontier Medicines' KRAS G12C-targeting program using chemoproteomics-informed machine learning; and numerous academic and industry programs targeting previously undruggable transcription factors, E3 ligases, and RNA-binding proteins through covalent mechanisms identified by ABPP.
The trend is clear: chemoproteomics is no longer a niche academic technique. It has become an essential engine of covalent drug discovery, integrated into the workflows of major pharmaceutical companies and venture-backed biotechnology startups alike. Creative Proteomics' Customized Experiments service supports chemoproteomics programs with tailored ABPP experimental design, from probe selection through quantitative MS acquisition and customized bioinformatics analysis.
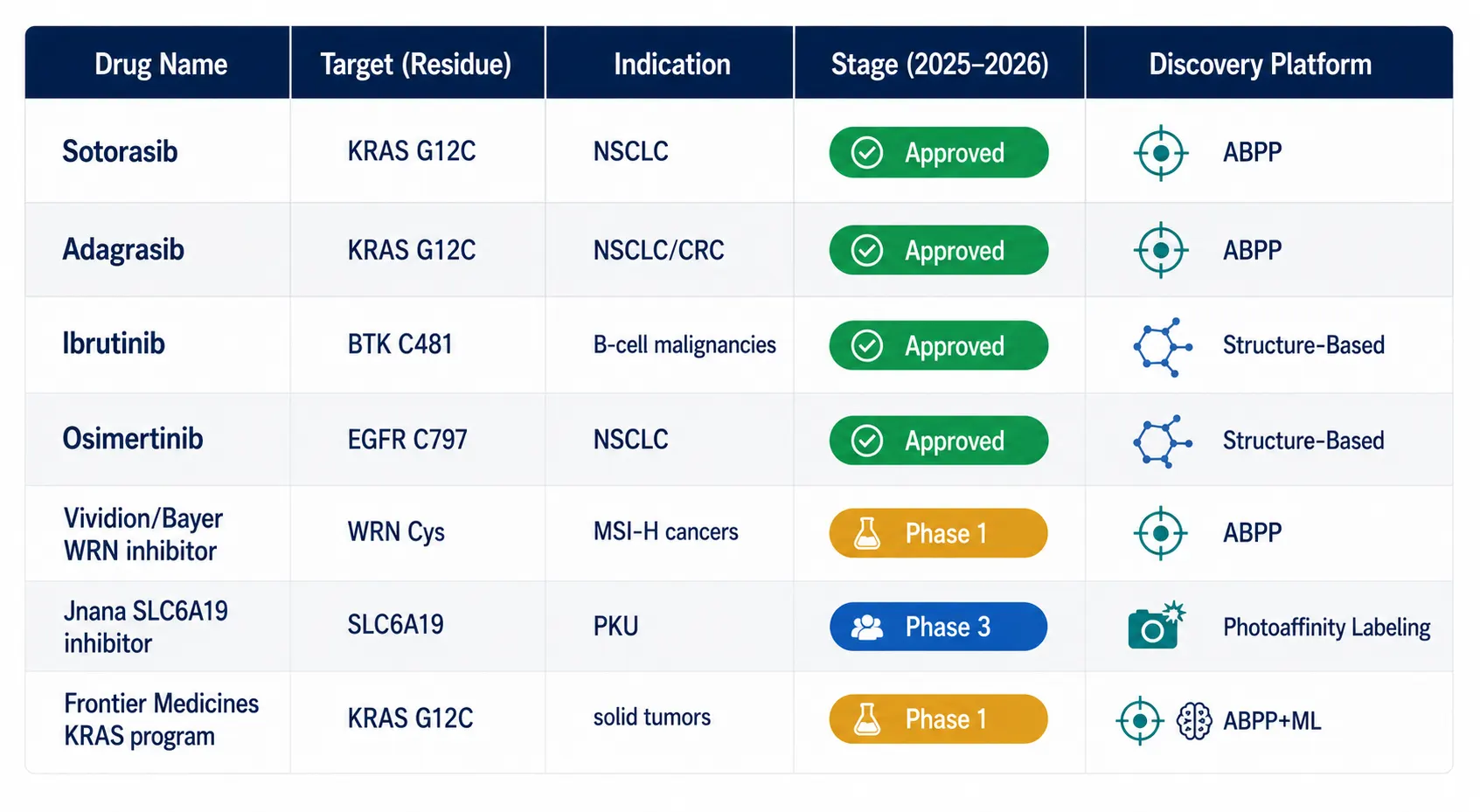 Figure 6: Clinical Covalent Inhibitor Pipeline
Figure 6: Clinical Covalent Inhibitor Pipeline
A clean modern table visualization showing the covalent inhibitor clinical pipeline as of 2025-2026, with drug names, targets, indications, stage (color-coded badges), and discovery platform, including sotorasib, adagrasib, ibrutinib, osimertinib, and emerging ABPP-discovered candidates.
FAQ
Q: What is the difference between ABPP and a regular pull-down experiment?
A regular pull-down uses an immobilized bait protein to capture interacting proteins. ABPP uses a small-molecule activity-based probe that covalently reacts with specific amino acid residues (usually cysteines) in their native protein context, then identifies the labeled proteins and the exact modified sites by mass spectrometry. ABPP provides site-level resolution that pull-downs cannot match.
Q: Why is cysteine so heavily favored over other amino acids for covalent inhibitors?
Cysteine's thiol group is the most nucleophilic side chain in proteins at physiological pH. This means it can selectively react with moderately electrophilic warheads like acrylamides, which are stable enough to survive in the bloodstream but reactive enough to engage their target. Other nucleophilic residues (lysine, histidine) require more reactive warheads that carry higher off-target risk.
Q: Can ABPP be used for reversible inhibitors, or only covalent ones?
ABPP directly detects covalent binding. For reversible inhibitors, modified versions of the technique — such as thermal proteome profiling (TPP-CETSA) or limited proteolysis-mass spectrometry (LiP-MS) — can detect target engagement through changes in protein thermal stability or protease susceptibility rather than direct covalent labeling. For additional insights into darts cetsa, explore our in-depth resource.
Q: How many proteins can ABPP profile in a single experiment?
A typical isoTOP-ABPP experiment can quantify 10,000-15,000 unique cysteine residues across 3,000-5,000 proteins in a human cell line. Coverage continues to improve with better mass spectrometers (timsTOF, Orbitrap Astral) and more efficient enrichment chemistries.
Q: How do I know if my covalent inhibitor is selective enough?
Competitive ABPP against the full proteome is the gold-standard selectivity assay. Run a dose-response ABPP experiment (multiple inhibitor concentrations, TMT-ABPP format) and calculate the competition ratio for every detected cysteine. A selective inhibitor should show strong competition (high ratio) at a handful of sites and minimal competition across the remaining >10,000 detected cysteines.
Q: What types of biological samples are compatible with ABPP experiments?
ABPP is compatible with cultured cell lysates, intact living cells (using cell-permeable probes), fresh or frozen tissue homogenates, and biofluid-derived proteomes such as plasma, serum, and cerebrospinal fluid. Sample preparation must preserve cysteine thiol redox status — fresh samples processed with thiol-protective buffers (e.g., containing tris(2-carboxyethyl)phosphine or N-ethylmaleimide to block post-lysis oxidation) yield the most reproducible labeling. For in vivo ABPP, tissues are harvested post-dosing and labeled ex vivo with the IA-alkyne probe.
References:
- Nuber CM, Schwab MA, Konrad DB. A perspective on cysteine-reactive activity-based probes. Organic & Biomolecular Chemistry. 2025;23(45):10262-10275. CC BY 3.0. doi:10.1039/D5OB00905G.
- Zhou YF, Zhang L, Niu ZL, Wang ZA. Targeting the reactive proteome: recent advances in activity-based protein profiling and probe design. Biomolecules. 2025;15(12):1699. CC BY 4.0. doi:10.3390/biom15121699.
- Schaefer D, Cheng X. Recent advances in covalent drug discovery. Pharmaceuticals. 2023;16(5):663. CC BY 4.0. doi:10.3390/ph16050663.
- Vitiello PP, Valsecchi AA, Duregon E, et al. KRAS G12C inhibition in solid tumors: biological breakthroughs, clinical evidence, and open challenges. Cancers. 2025;17(17):2803. CC BY 4.0. doi:10.3390/cancers17172803.
- Miyashita H, Kato S, Hong DS. KRAS G12C inhibitor combination therapies: current evidence and challenge. Frontiers in Oncology. 2024;14:1380584. CC BY 4.0. doi:10.3389/fonc.2024.1380584.













