Identifying what proteins a drug actually binds inside a cell is one of the hardest problems in drug discovery. A compound may show potent activity in a phenotypic screen, but without knowing its molecular target, medicinal chemists cannot optimize binding affinity, understand selectivity, or anticipate toxicity. Chemoproteomics — the intersection of chemical biology and quantitative mass spectrometry — has emerged as the definitive toolkit for solving this problem. Rather than relying on a single method, the modern target deconvolution workflow integrates probe-based enrichment strategies with probe-free biophysical profiling to triangulate drug-target interactions from multiple independent angles. This article provides a practical decision framework for selecting and combining these approaches, with emphasis on how each method contributes distinct evidence to the target identification cascade.
The Target Deconvolution Challenge
Target deconvolution — tracing a phenotypic hit back to its molecular target — remains the rate-limiting step in phenotypic drug discovery. Unlike target-based screening, where the protein is known from the start, phenotypic campaigns begin with a cellular or organismal readout and work backward. The challenge is fundamentally a needle-in-a-haystack problem: any given small molecule can interact with dozens or hundreds of proteins across the proteome, yet only one or a few of these interactions are responsible for the observed biological activity.
Several factors compound the difficulty. First, drug-target interactions span an enormous affinity range, from sub-nanomolar covalent adducts to millimolar transient contacts, and no single method captures this full spectrum equally well. Second, the biologically relevant target may be of low abundance, making it invisible to methods that lack an enrichment step. Third, drug binding often induces conformational changes or engages protein complexes rather than isolated polypeptides, requiring methods that preserve native structure. Fourth, many bioactive compounds — particularly natural products — lack convenient synthetic handles for chemical modification, ruling out probe-based approaches that require derivatization.
These constraints mean that effective target deconvolution is rarely a one-experiment endeavor. Instead, it requires a staged strategy in which orthogonal methods provide complementary lines of evidence: one method screens broadly to generate candidate lists, a second confirms direct engagement, and a third maps the binding site at amino acid resolution. The following sections walk through each major category of chemoproteomic methods, their strengths and blind spots, and how they fit into this staged framework.
Probe-Based Methods: ABPP, Affinity Pulldown, and Photoaffinity Labeling
Probe-based chemoproteomics uses a chemically modified version of the drug — typically outfitted with an alkyne or biotin handle — to physically retrieve target proteins from a complex lysate. The core advantage is enrichment: by pulling down the drug-protein complex, even low-abundance targets become detectable by LC-MS/MS. This category encompasses three principal approaches.
Activity-Based Protein Profiling (ABPP) uses probes designed to react covalently with the active-site residues of specific enzyme classes. A typical ABPP probe contains a reactive warhead (e.g., an iodoacetamide for cysteine, a fluorophosphonate for serine hydrolases), a flexible linker, and a click-chemistry handle. After incubating the probe with a cell lysate or live cells, copper-catalyzed azide-alkyne cycloaddition (CuAAC) attaches a biotin tag, enabling streptavidin enrichment of probe-labeled proteins. Competitive ABPP extends this concept: by pre-incubating samples with an unmodified inhibitor before adding a broad-spectrum activity probe, proteins whose active sites are protected by the inhibitor show reduced probe labeling. This competitive format enables dose-response profiling across hundreds of cysteine or serine residues simultaneously, revealing both the primary target and off-targets. Creative Proteomics offers TMT-based proteomics services that integrate directly with competitive ABPP workflows, providing multiplexed quantification across up to 18 treatment conditions in a single LC-MS/MS run.
For researchers who prefer to outsource the computational workflow, Creative Proteomics' bioinformatics for proteomics platform handles raw file processing through statistical hit calling to pathway enrichment analysis, with customizable pipelines for each chemoproteomic method.
Affinity pulldown is the simplest probe-based method: the compound of interest is immobilized on a solid support (e.g., agarose or magnetic beads) via a linker, incubated with a cell lysate, and washed to remove non-specific binders before elution and MS analysis. The method is straightforward but suffers from two well-known limitations. First, linker attachment can sterically block the compound's protein-binding surface, producing false negatives. Second, abundant proteins with non-specific bead affinity — so-called "sticky" proteins — routinely appear as false positives. Competitive elution with excess free compound helps distinguish specific from non-specific interactions.
Photoaffinity labeling (PAL) addresses the limitation of affinity pulldown for weak or transient binders. A photoreactive group (diazirine, benzophenone, or aryl azide) is incorporated into the compound, which upon UV irradiation generates a highly reactive carbene or nitrene that crosslinks to nearby amino acid side chains irrespective of binding affinity. This covalent capture step is performed in living cells before lysis, preserving the native cellular context. After crosslinking, a click-chemistry handle is appended for enrichment. PAL is particularly valuable for natural products and fragment-sized molecules whose binding is too weak to survive the washing steps of traditional pulldown.
Probe-Free Methods: TPP-CETSA, LiP-MS, DARTS, and SPROX
Not every compound can be chemically modified without losing activity. Natural products with dense stereochemistry, fragments with few functional groups, and compounds whose structure-activity relationship is extremely tight all pose challenges for probe design. Probe-free methods — also called stability-based or biophysical chemoproteomics — exploit the thermodynamic and structural consequences of drug binding without requiring any compound derivatization. For additional insights into darts cetsa, explore our in-depth resource.
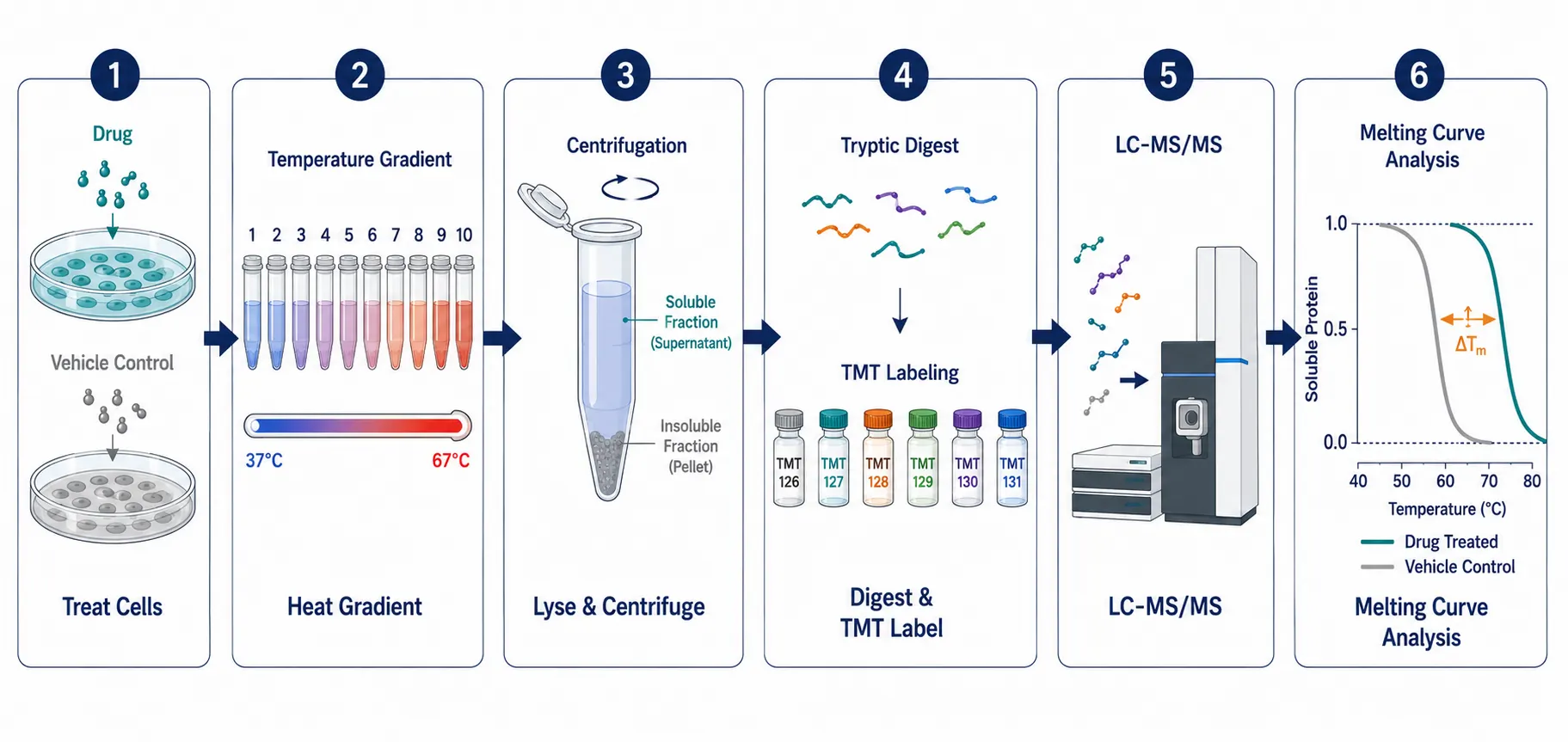
Thermal Proteome Profiling (TPP), also known as the cellular thermal shift assay coupled to mass spectrometry (CETSA-MS), is the most widely adopted probe-free platform. The principle is elegant: when a protein binds a ligand, its thermal stability increases — it unfolds and precipitates at a higher temperature than the unbound form. In a TPP experiment, drug-treated and vehicle-treated cells or lysates are aliquoted across a temperature gradient (typically 37-67 degrees C, 10 points), heated briefly, and centrifuged to remove precipitated proteins. The soluble fraction at each temperature is digested and analyzed by quantitative LC-MS/MS (Figure 3). Proteins that show a rightward shift in their melting curve (a positive delta-Tm) in the drug-treated sample are candidate targets. The method is proteome-wide — a single TPP experiment can monitor thermal stability changes for 5,000-8,000 proteins — and works in intact cells, lysates, and even tissue samples. Because no probe is required, TPP captures direct binding events as well as downstream consequences of target engagement, such as the recruitment of a protein to a stabilized complex.
Limited Proteolysis Mass Spectrometry (LiP-MS) provides a complementary readout: instead of thermal stability, it measures conformational changes induced by drug binding. A drug-bound protein adopts a different structural state than its unbound form, which alters the accessibility of protease cleavage sites. In LiP-MS, a drug-treated proteome is subjected to brief, limited proteolysis (typically with proteinase K), and the resulting peptide fragments are quantified by LC-MS/MS. Peptides whose abundance changes significantly between drug-treated and control samples pinpoint regions of the protein that underwent a binding-induced conformational change — effectively mapping the binding site to within approximately 12 amino acids. LiP-MS is uniquely capable of detecting binding events for metabolites, cofactors, and other non-covalent ligands, and it works in complex lysates without any compound modification.
Drug Affinity Responsive Target Stability (DARTS) is the simplest and most accessible probe-free method. It relies on the observation that drug binding protects a target protein from proteolysis. A cell lysate is incubated with the drug, then subjected to limited digestion with a non-specific protease such as pronase or thermolysin. Proteins that are stabilized by drug binding persist longer in the digestion, appearing as protected bands on an SDS-PAGE gel relative to the vehicle control. These bands are excised and identified by MS. For laboratories that lack in-house MS capabilities, Creative Proteomics' protein identification services provide complete target protein characterization from excised gel bands, including sequence coverage analysis and post-translational modification mapping. DARTS requires no chemical modification, works with crude lysates, and can be performed with standard biochemistry equipment, making it an attractive first-pass screening tool. Its main limitation is low proteome coverage: abundant proteins dominate the gel, and low-abundance targets are easily missed.
Stability of Proteins from Rates of Oxidation (SPROX) rounds out the probe-free toolkit. Rather than heat or proteolysis, SPROX uses chemical denaturation: proteins are exposed to increasing concentrations of a chaotropic denaturant (guanidinium hydrochloride or urea) in the presence of hydrogen peroxide, which oxidizes methionine residues in unfolded but not folded proteins. The extent of methionine oxidation at each denaturant concentration is quantified by MS, generating a denaturation curve. Ligand-bound proteins resist denaturation, producing a rightward shift. SPROX is methionine-dependent — proteins lacking methionine are invisible — but it provides a genuinely orthogonal physical principle that complements thermal and proteolytic methods.
Method Comparison Matrix
The table and figure below provide a side-by-side comparison of the five major chemoproteomic target ID methods to guide initial method selection (Figure 1, Table 1). All methods listed are available through Creative Proteomics' proteomics service platform.
Figure 1: Chemoproteomic Target Deconvolution Method Comparison Matrix

| Method | Chemical Modification Required | Proteome Coverage | Binding Site Resolution | Live-Cell Compatible | Best Application |
|---|---|---|---|---|---|
| ABPP | Yes (probe synthesis) | Medium-High (enzyme families) | Active-site residue level | Yes (cell-permeable probes) | Covalent inhibitors, enzyme targets, competitive dose-response profiling |
| Affinity Pulldown | Yes (immobilization) | Low-Medium (bead-dependent) | Protein-level only | No (lysate only) | High-affinity binders (Kd < 1 microM), initial screening |
| Photoaffinity Labeling | Yes (photoreactive group) | Medium | Crosslink site | Yes (UV in live cells) | Weak/transient binders, natural products, fragments |
| TPP/CETSA-MS | No | High (5,000-8,000 proteins) | Protein-level only | Yes (intact cells or tissues) | Unbiased proteome-wide screening, polypharmacology profiling, in vivo target engagement |
| LiP-MS | No | Medium (dependent on sequence coverage) | Approximately 12 amino acid precision | No (lysate only) | Binding site mapping, conformational change detection, non-covalent ligands |
| DARTS | No | Low (<1,000 proteins) | Protein-level only | No (lysate only) | Low-cost pilot screen, natural products, orthogonal validation |
| SPROX | No | Low-Medium (Met-dependent) | Protein-level only | No (lysate only) | Orthogonal validation, complementing TPP data with thermodynamic readout |
Quantitative Mass Spectrometry for Target Identification
Regardless of which chemoproteomic method generates the samples, the back end is always quantitative LC-MS/MS. The choice of quantification strategy — TMT multiplexing, label-free quantification (LFQ), or data-independent acquisition (DIA) — has profound effects on throughput, quantitative accuracy, and statistical power.
TMT-based quantification is the workhorse of chemoproteomic target ID. Tandem mass tags are isobaric labels that allow up to 18 samples (TMTpro 18-plex) to be pooled and analyzed simultaneously in a single MS run. In a TPP experiment, each temperature point in the gradient receives a different TMT channel, and reporter ion intensities from MS2 or MS3 fragmentation provide relative protein abundance at each temperature. The multiplexing eliminates run-to-run variation and reduces missing values — a critical advantage when fitting melting curves that require complete data across all temperature points. Creative Proteomics' TMT-based proteomics service supports up to 18-plex experimental designs with SPS-MS3 quantification for accurate reporter ion ratios.
Label-free quantification (LFQ) trades multiplexing for simplicity and dynamic range. Samples are run individually, and peptide intensities are compared across runs using extracted ion chromatograms. LFQ is well-suited to DARTS and affinity pulldown experiments where the sample number is small and the goal is fold-change comparison between drug-treated and control. Creative Proteomics' label-free quantification service supports both DDA and DIA acquisition strategies, with optimized data processing pipelines that minimize missing values across large sample cohorts.
Data-independent acquisition (DIA) represents a paradigm shift in quantitative proteomics. Unlike data-dependent acquisition (DDA), where the mass spectrometer selects the top-N most abundant precursor ions for fragmentation, DIA systematically fragments all precursors within defined m/z windows regardless of abundance. This generates a complete digital record of all ionizable peptides in the sample, which is then mined against a spectral library. The advantages for chemoproteomics are compelling: DIA eliminates the stochastic precursor selection of DDA, dramatically reducing missing values across temperature points or treatment conditions; it provides more consistent quantification of low-abundance peptides, improving sensitivity for low-abundance targets; and the same DIA data can be re-analyzed with updated libraries as new target hypotheses emerge. Creative Proteomics' DIA quantitative proteomics service leverages high-resolution Orbitrap instruments for deep proteome coverage with excellent quantitative reproducibility.
Statistical Hit Calling: NPARC and Beyond
Identifying drug targets from chemoproteomic data is ultimately a statistical problem. In a TPP experiment with 5,000 quantified proteins, how do you distinguish the handful of genuine targets from random thermal stability fluctuations?
The most commonly used framework is non-parametric analysis of response curves (NPARC), implemented as an R/Bioconductor package. NPARC fits two models to each protein's melting curve: a null model (a single sigmoid through both drug-treated and vehicle data points) and an alternative model (separate sigmoids for drug and vehicle). An F-test compares the goodness of fit between the two models, and proteins for which the alternative model explains the data significantly better are called as hits. NPARC makes no assumptions about the shape of the melting curve beyond monotonicity, making it robust across diverse protein behaviors.
A 2025 benchmarking study in Molecular & Cellular Proteomics compared NPARC against MSstatsTMT, a newer framework specifically designed for TMT-based quantitative proteomics. The key finding was that MSstatsTMT, which models all sources of variation within a unified linear mixed-effects framework, detected more true positives at equivalent false discovery rates than NPARC when applied to TPP data. MSstatsTMT also supports flexible experimental designs — including the "OnePot" format in which multiple temperature points are pooled before TMT labeling to reduce sample handling. Regardless of the specific algorithm chosen, the consensus is that statistical hit calling should incorporate biological replicates (at least three), control for protein abundance (higher-abundance proteins show artificially stable melting curves), and be followed by orthogonal validation with an independent method. Creative Proteomics' statistical analysis service provides dedicated support for chemoproteomic data processing, including NPARC and MSstatsTMT implementation, multiple testing correction, and visualization of melting curve shifts across the proteome.
Method Selection Framework
Choosing the right target deconvolution strategy is context-dependent. The decision tree below (Figure 2) integrates the key considerations — compound properties, prior knowledge, available resources, and desired resolution — into a practical workflow.
Figure 2: Method Selection Decision Tree for Drug-Target Identification
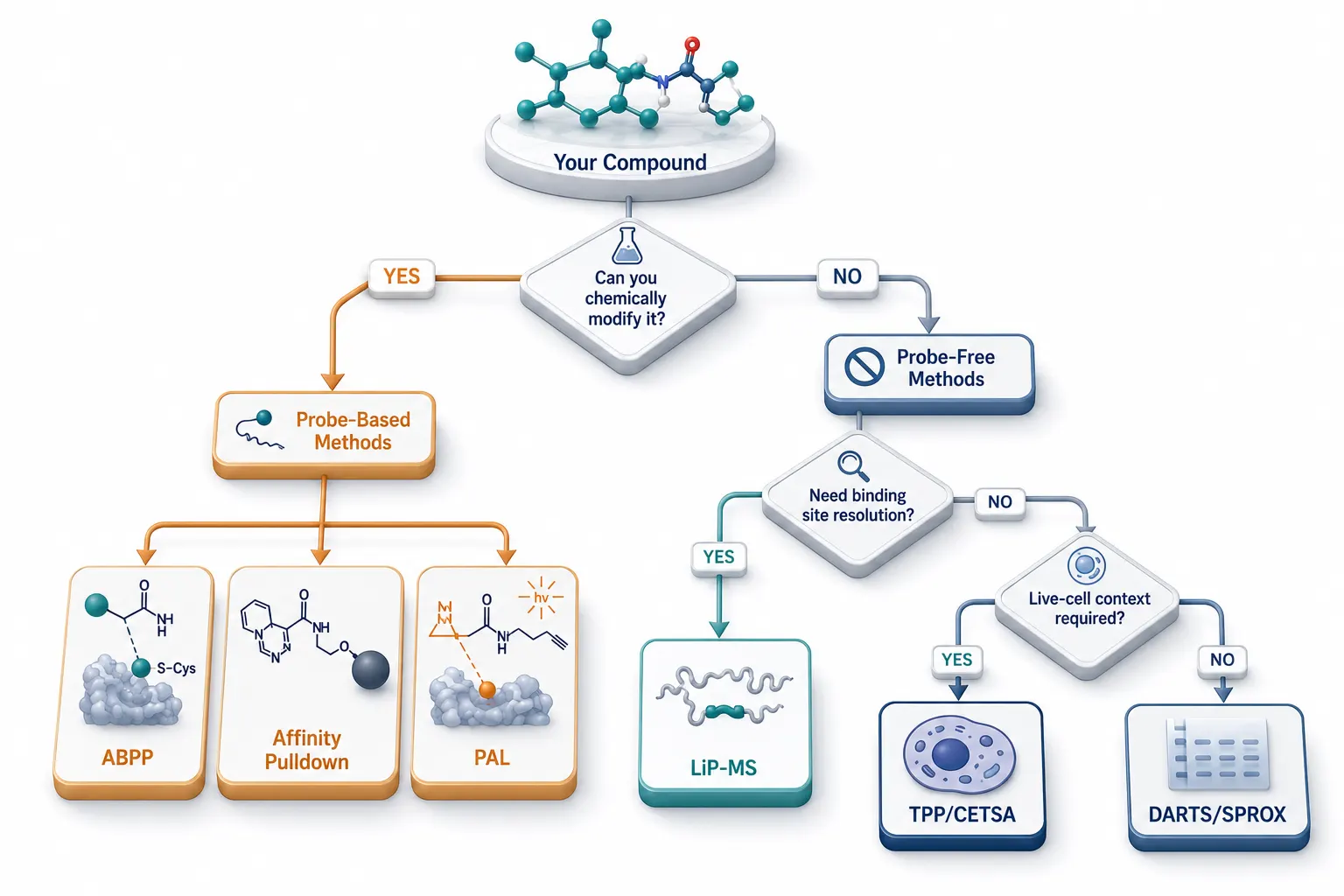
Start with the compound. If it contains a reactive electrophile (e.g., an acrylamide, chloroacetamide, or sulfonyl fluoride), or if structure-activity relationship data indicate a covalent mechanism, competitive ABPP is the first-line approach. It provides site-specific target identification, built-in selectivity profiling, and dose-response information in a single experiment. If the compound is not a covalent inhibitor but has a tractable synthetic handle for derivatization, consider PAL or affinity pulldown. PAL is preferred for weak binders where the crosslinking step captures transient interactions that would be lost during pulldown washes.
If chemical modification is impossible — the classic scenario for natural products, fragments, and compounds in early hit-to-lead optimization — start with a probe-free method. TPP is the most broadly applicable first-pass screen, providing proteome-wide coverage in live cells. For academic labs or pilot studies with limited MS access, DARTS offers a rapid, low-cost entry point. For projects where the binding site must be identified (e.g., to guide medicinal chemistry), LiP-MS adds structural resolution that neither TPP nor DARTS can provide.
In practice, the most robust deconvolution campaigns combine methods. A common workflow is: TPP for unbiased primary screening, then CETSA-WB on the top 5-10 hits for orthogonal confirmation, then competitive ABPP or LiP-MS for binding site resolution, then SPR or BLI with purified recombinant protein for affinity quantification. Each stage adds a distinct type of evidence, and no single experiment is treated as dispositive on its own.
Integrated Case Studies: ABPP + TPP in Practice
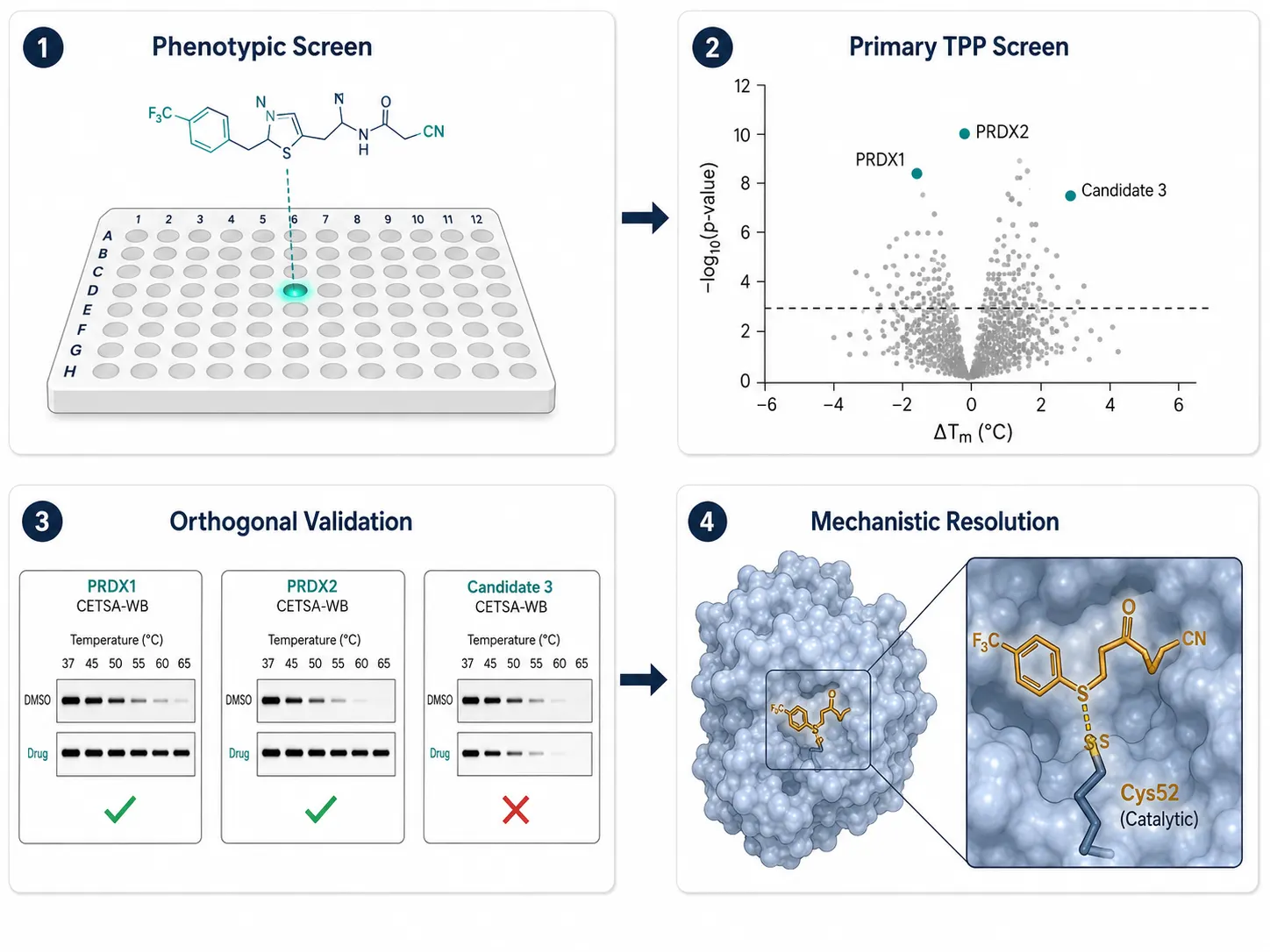
The power of multi-method integration is best illustrated by real discovery campaigns (Figure 4).
The KRAS G12C story, culminating in the FDA approvals of sotorasib (2021) and adagrasib (2022), relied heavily on chemoproteomic target engagement data. Early fragment screens identified compounds that bound covalently to the mutant cysteine-12 in the switch-II pocket, a site previously considered undruggable. Competitive isoTOP-ABPP experiments confirmed that these fragments engaged KRAS G12C selectively across the proteome, with minimal off-target labeling. Later, TPP experiments in KRAS-mutant cell lines showed that sotorasib stabilized KRAS G12C against thermal denaturation, providing orthogonal evidence of target engagement in living cells. The combination of ABPP (for site-specific selectivity) and TPP (for cellular target engagement) was instrumental in building the data package that supported clinical development.
In the natural product space, the sesquiterpene lactone ainsliadimer A was found to inhibit NF-kB signaling in cancer cells, but its protein target was unknown. A 2023 study deployed a three-method cascade: DARTS first identified two peroxiredoxin family members (PRDX1 and PRDX2) as stabilized bands on SDS-PAGE. TPP with a temperature-range design (TPP-TR) confirmed that both PRDX1 and PRDX2 showed significant thermal stabilization in the presence of ainsliadimer A across the proteome. Finally, CETSA-WB with PRDX1- and PRDX2-specific antibodies validated the interaction in living cells, and site-directed mutagenesis of the catalytic cysteines abolished binding, confirming that ainsliadimer A acts through covalent modification of the peroxiredoxin active site. This study illustrates the archetypal deconvolution cascade: unbiased screen, proteome-wide confirmation, targeted validation, mechanistic resolution.
A third instructive case comes from traditional Chinese medicine. Curcumol, a guaiane-type sesquiterpene isolated from Curcuma species, was known to exhibit anticancer activity but its direct protein target remained unknown. A CETSA-based target identification campaign identified nucleolin as the specific binding target of curcumol in cancer cells, with the drug-induced thermal stabilization confirmed by both WB-CETSA and TPP. Functional validation demonstrated that curcumol binding to nucleolin disrupts rRNA synthesis and inhibits ribosome biogenesis, directly explaining the compound's antiproliferative effects. Notably, curcumol is a small, non-covalent natural product with no synthetic handle — it would have been completely invisible to traditional probe-based chemoproteomic methods. This case underscores why thermal stability-based approaches are essential components of the target deconvolution toolkit.
Figure 3: TPP-CETSA Experimental Workflow Pipeline
Validation Cascade: From Hit to Confirmed Target
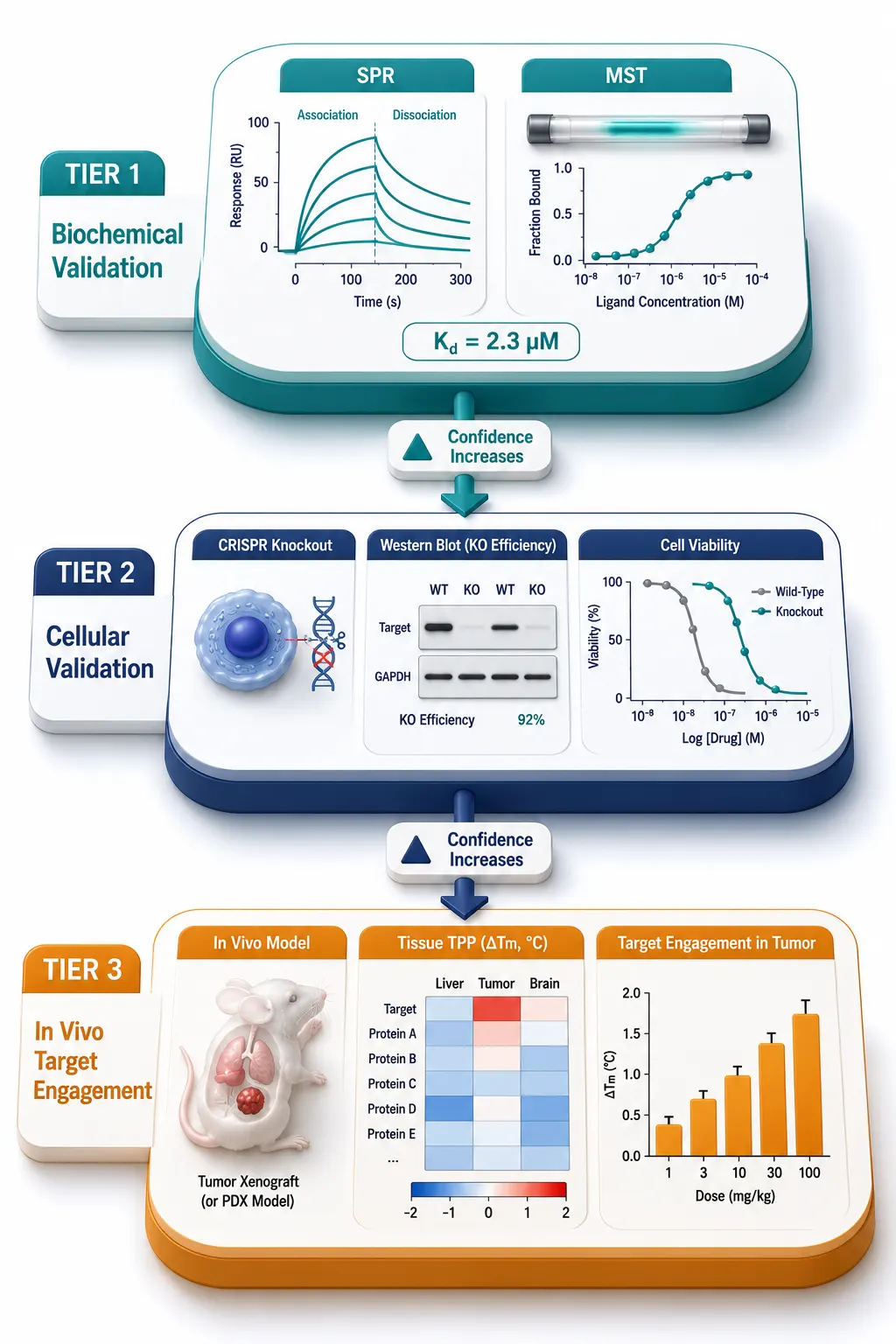
A list of candidate targets from a chemoproteomic screen is a starting point, not a conclusion. The validation cascade (Figure 5) moves candidates through increasingly stringent tests of biological relevance.
The first filter is orthogonal target engagement. A protein identified by TPP should be confirmed by CETSA-WB, SPR, or MST with purified recombinant protein. If the hit came from ABPP, a label-free method such as DARTS or LiP-MS provides orthogonal physical evidence that does not depend on the same probe chemistry. The principle is that two methods exploiting different physical properties — thermal stability vs. proteolytic susceptibility, for instance — are unlikely to produce the same false positive.
The second filter is functional relevance. siRNA knockdown or CRISPR/Cas9 knockout of the candidate target should phenocopy the compound's effect. If the compound inhibits cell proliferation with an IC50 of 2 microM, knockdown of the target should similarly reduce proliferation. Conversely, overexpression of the target should confer resistance. For covalent inhibitors, a mutation at the reactive residue (e.g., Cys-to-Ser or Cys-to-Ala) should abolish both labeling and biological activity, establishing a direct causal link between target modification and pharmacology.
The third and most demanding filter is in vivo target engagement. It is one thing to show that a compound binds its target in a cell lysate; it is quite another to demonstrate binding in an animal model at a therapeutically relevant dose. Tissue-TPP and blood-CETSA have recently emerged as methods for measuring target engagement directly in animal tissues. In tissue-TPP, drug-treated mice are sacrificed, organs are harvested, and tissues are subjected to the same thermal profiling workflow used for cell lysates. A dose-dependent thermal stabilization of the target in liver, tumor, or brain tissue provides direct pharmacokinetic-pharmacodynamic (PK-PD) linkage. For covalent inhibitors, click-chemistry-based pull-down from tissue lysates followed by Western blot or targeted MS offers a simpler alternative. Creative Proteomics' protein-protein interaction analysis service supports orthogonal target engagement validation by confirming whether drug binding alters the target's interaction network via co-immunoprecipitation-MS and chemical cross-linking mass spectrometry.
Figure 4: Integrated Case Study — From Phenotypic Hit to Validated Target
Emerging Frontiers: In Vivo, Single-Cell, and AI-Driven Target ID
The chemoproteomic target identification landscape continues to evolve rapidly. Three developments deserve particular attention.
Tissue-clearing chemoproteomics represents a step change in spatial resolution. The vCATCH (volumetric Clearing and Analysis of Target engagement by CHemoproteomics) method, published in 2025, uses solvent-based tissue clearing to render intact mouse organs optically transparent while preserving covalent drug-target adducts. After clearing, click chemistry is performed in situ to attach fluorescent reporters, and light-sheet microscopy maps the three-dimensional distribution of target engagement across entire organs at single-cell resolution. Applied to the BTK inhibitor ibrutinib, vCATCH revealed that the drug engages its target not only in the expected B-cell compartments of the spleen but also in unexpected regions of the brain, identifying a previously unrecognized tissue distribution pattern.
At the opposite end of the spatial scale, single-cell thermal profiling is pushing CETSA toward cellular resolution. By combining fluorescence-activated cell sorting with thermal challenge, researchers can now measure target engagement in specific cell populations within a heterogeneous tissue. This is critical for oncology, where a drug may engage its target in tumor cells but not in stromal or immune cells, with profound implications for efficacy and toxicity.
AI-driven target prediction is perhaps the most transformative trend. Machine learning models trained on large-scale chemoproteomic datasets can now predict which proteins a compound is likely to bind based on its chemical structure alone, allowing researchers to prioritize experimental validation. AlphaFold-predicted structures are increasingly used to identify cryptic binding pockets that are invisible to traditional structural biology, and molecular dynamics simulations can rank candidate targets by predicted binding free energy. For Creative Proteomics clients, these computational predictions can be directly coupled with experimental validation — an AI-predicted target list is fed into a TPP or ABPP workflow, creating a closed loop between in silico prediction and bench-top confirmation. Creative Proteomics' customized experiments service allows researchers to design bespoke chemoproteomic workflows that combine any of the methods discussed in this article — ABPP, TPP, LiP-MS, or PAL — within a single integrated project, from primary screening through to validated target.
Figure 5: Target Validation Cascade — Biochemical to In Vivo
Frequently Asked Questions
What is the difference between ABPP and TPP for target identification?
ABPP uses a chemically modified activity-based probe to covalently label and enrich specific enzyme classes, providing residue-level target identification. TPP requires no probe modification and monitors thermal stability shifts across the entire proteome, providing unbiased coverage but protein-level resolution only. The two methods are complementary and are often used together in integrated workflows.
Can chemoproteomics identify targets of non-covalent drugs?
Yes. TPP, LiP-MS, DARTS, and SPROX all work with non-covalent ligands, as they detect binding through changes in protein physical properties (thermal stability, protease susceptibility, chemical denaturation resistance) rather than covalent bond formation. Photoaffinity labeling can also capture non-covalent interactions through UV-induced crosslinking.
How many proteins can TPP detect in a single experiment?
A typical TPP experiment quantifies 5,000-8,000 proteins across the temperature gradient, depending on the sample complexity, MS instrument sensitivity, and fractionation depth. The latest Orbitrap Astral instruments can push coverage beyond 10,000 proteins.
What statistical method is best for analyzing TPP data?
Both NPARC (non-parametric analysis of response curves) and MSstatsTMT are widely used. MSstatsTMT, published in 2025, provides a unified linear mixed-effects framework that handles both traditional multi-temperature and pooled OnePot experimental designs and is recommended for new TPP users.
Do I need to modify my compound for chemoproteomic target identification?
Not necessarily. Probe-free methods (TPP, LiP-MS, DARTS, SPROX) work with unmodified compounds. If you can modify your compound, probe-based methods (ABPP, PAL, affinity pulldown) provide higher sensitivity through target enrichment.
How long does a typical target deconvolution campaign take?
A primary TPP or ABPP screen can be completed in 2-4 weeks. Full validation — including orthogonal confirmation, functional studies, and affinity measurements — typically requires 3-6 months depending on the number of candidates and the complexity of the biological system.
References:
- Zou M, Zhou H, Gu L, Zhang J, Fang L. Therapeutic target identification and drug discovery driven by chemical proteomics. Biology. 2024;13(8):555. CC BY 4.0. https://doi.org/10.3390/biology13080555
- Du P, Fan R, Zhang N, Wu C, Zhang Y. Advances in integrated multi-omics analysis for drug-target identification. Biomolecules. 2024;14(6):692. CC BY 4.0. https://doi.org/10.3390/biom14060692
- Song J. Applications of the Cellular Thermal Shift Assay to Drug Discovery in Natural Products: A Review. International Journal of Molecular Sciences. 2025;26(9):3940. CC BY 4.0. https://doi.org/10.3390/ijms26093940
- Mons E, Kim RQ, Mulder MPC. Technologies for direct detection of covalent protein-drug adducts. Pharmaceuticals. 2023;16(4):547. CC BY 4.0. https://doi.org/10.3390/ph16040547
- Figueroa-Navedo AM, Kapre R, Gupta T, et al. MSstatsTMT improves accuracy of thermal proteome profiling by trading off temperature treatments and biological replicates. Molecular & Cellular Proteomics. 2025;24(8):100999. CC BY 4.0. https://doi.org/10.1016/j.mcpro.2025.100999














